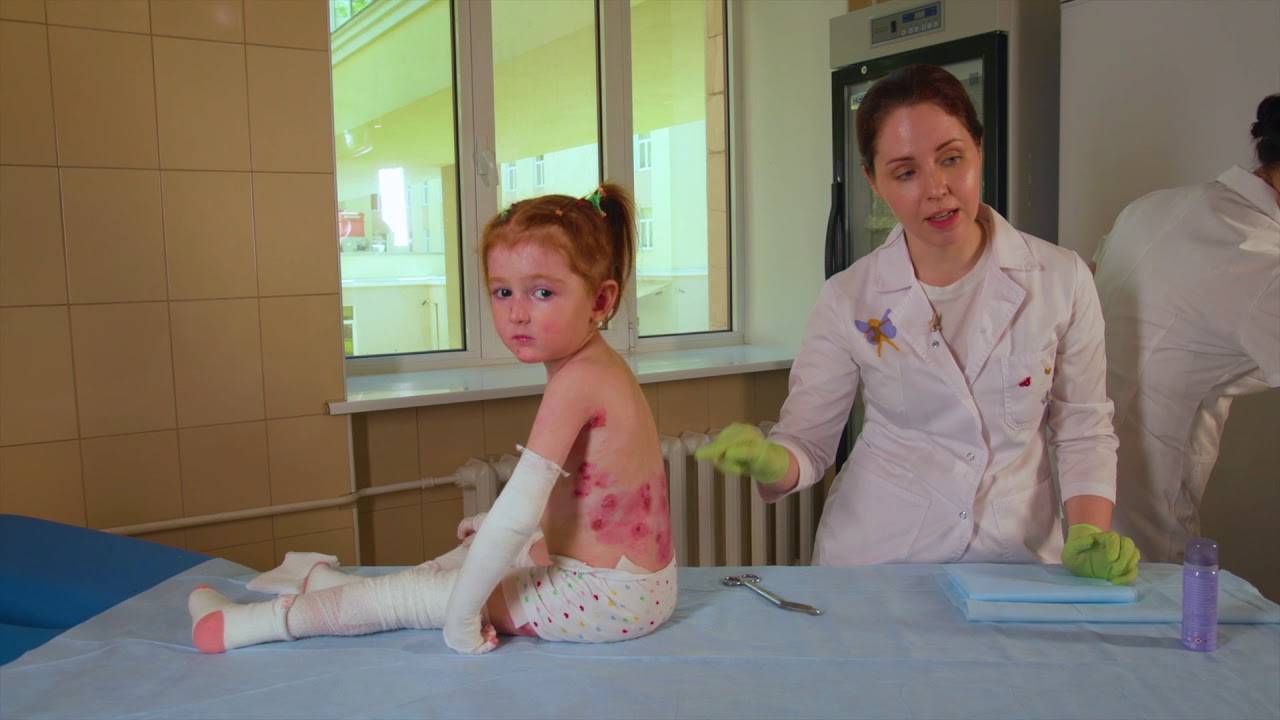
Введение – буллезный эпидермолиз, что это?
Буллезный эпидермолиз – это редкое хроническое наследственное заболевание, главный его признак – образование пузырей и мокнущих ран (эрозий) на коже и слизистых оболочках, возникающих при незначительном травмировании. Заболевание заметно сразу после рождения или в первые месяцы жизни ребенка. Существуют как тяжелые формы, при которых дети умирают в течение нескольких дней или месяцев после рождения от инфекционных осложнений или становятся инвалидами, так и легкие формы, при которых человек может вести полноценную жизнь при проведении соответствующего лечения.
При некоторых формах буллезного эпидермолиза бывают поражены только небольшие участки кожи, при других – большие по площади участки поражения, на всей коже, часто с вовлечением слизистых оболочек и ногтей. Определенные виды буллезного эпидермолиза вызывают поражение внутренних органов. Наиболее часто страдает желудочно-кишечный тракт в виде непроходимости пищевода, нарушения глотания, сужения пищевода, поносов, запоров, поражений прямой кишки, желудка, ногтей, сращение пальцев, сердца (кардиомиопатия), мышц (дистрофия), глаз (выворот век), повреждение роговицы, зубов (кариес, плохая эмаль) и пр.
Данное заболевание встречается редко. Так, за период 1986-1990 года в Соединенных Штатах было зарегистрировано 19 случаев на 1 млн. новорожденных. Но, несмотря на редкость его возникновения, оно требует активной диагностики, лечения, наблюдения.
Этиология
Буллезный эпидермолиз (БЭ) – название группы генетических заболеваний, характеризующихся чрезмерной восприимчивостью кожи и слизистых оболочек к отделению от более глубоких тканей в результате механических повреждений . БЭ может быть аутосомно-рецессивным или аутосомно-доминантным; в целом, рецессивные формы более серьезны. Более 1000 зарегистрированных мутаций в 14 генах влияют на различные формы БЭ, что приводит к проявлению огромного разнообразия клинических случаев БЭ. .
Четыре типа БЭ в основном классифицируются по уровню расщепления дермо-эпидермальных соединений :
- Простой буллезный эпидермолиз (ПБЭ) – наиболее мягкая и распространенная форма. Простой БЭ вызывает появление волдырей в местах трения, обычно поражая руки и ноги. Он поражает гены кератинов эпидермиса.
- Пограничный буллезный эпидермолиз (ПБЭ) проявляется при рождении и обычно более серьезен, чем простой БЭ. Пограничный БЭ выражается появлением волдырей в местах трения. Этот тип заболевания поражает ламинин и коллаген.
- Дистрофический буллезный эпидермолиз (ДБЭ) может быть как легким, так и тяжелым, он поражает кожу и другие органы. Дистрофический БЭ вызывается генетическими мутациями, повреждающими производство коллагена. При тяжелых формах ДБЭ возникает множество осложнений, влияющих на способность пациента к выздоровлению. Основными осложнениями являются недоедание, анемия, трудно поддающийся лечению зуд, боль, инфекция и угрожающая колонизация.
- Синдром Киндлера – редкое трудно диагностируемое заболевание БЭ, которое часто путают с другими подтипами БЭ. Вызывается мутациями гена FERMT1. В результате мутаций гена FERMT1 появляются волдыри, эпидермальная атрофия и задержка выздоровления. Травматические кожные волдыри появляются в раннем детстве и преобладают наряду с потерей кожи и ранениями в неонатальном периоде.
Сколько людей болеют буллёзным эпидермолизом?
Чаще всего недуг регистрируется у детишек возраста 1 — 5 лет. Данные Национального регистра больных буллёзным эпидермолизом, который ведётся в Соединённых Штатах Америки, говорят о том, что недугом страдает 1 из 50000 новорождённых. За 16 лет его существования в США выявлены 3300 человек с данной болезнью.
В Европе буллёзным эпидермолизом страдает 1 из 30000 новорождённых малышей. В Японии наименьшая распространённость болезни, она выявляется у 7,8 из 1 млн рождённых детей.
К сожалению, в России отсутствует официальная статистика по заболеванию. Известно, что в нашей стране, Украине, Белоруссии и Казахстане насчитывается более 150 пациентов с буллёзным эпидермолизом.
Диагностика
В зависимости от периода проведения диагностических мероприятий их можно подразделить на пренатальные (до рождения ребенка) и постнатальные (после рождения ребенка).
Существует два основных метода постнатальной диагностики буллезного эпидермолиза – иммунофлюоресцентное генетическое картирование (IAM) и электронная микроскопия (TEM).
Для осуществления этих видов исследований у пациента осуществляется забор образцов кожи. Для проведения IAM и TEM образцы кожи получают при помощи биопсии. Осуществляют забор неповрежденного и поврежденного участков кожи. При IAM определяют наличие специфических белков кожи. Для этого используют специальные моноклональные антитела, которые избирательно связываются только с определенным белком кожи. Если данный белок отсутствует, то моноклональные антитела остаются несвязанными, окрашивания специальным светящимся составом не происходит.
При электронной микроскопии определяются конкретные компоненты кожи, отсутствие которых способно вызвать развитие буллезного эпидермолиза – кератиновые филаменты, полудесмосомы, якорные фибриллы и пр.
Также значительную роль играет анализ ДНК, проводимый с целью выявления генетической мутации и способа наследования. Но данный метод исследования используется только при наличии изменений, найденных при IAM и TEM, так как анализу необходимо подвергнуть слишком много генов, которые могут быть повреждены.
В пренатальной диагностике используется материал, полученный из околоплодных вод при сроке беременности более 17 недель. Поврежденный ген определяется путем анализа ДНК.
Осложнения буллёзного эпидермолиза
Развитие осложнений чаще развиваются у детишек, страдающих пограничным и дистрофическим буллёзным эпидермолизом. Болезни этих групп протекают в самых агрессивных формах.
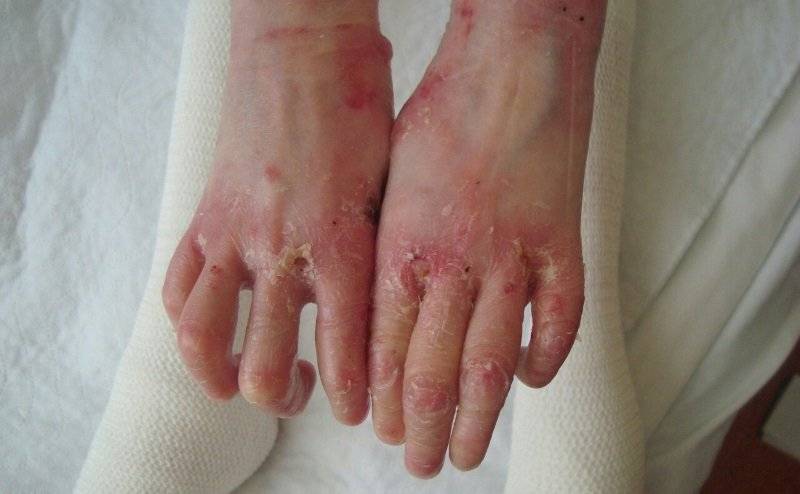
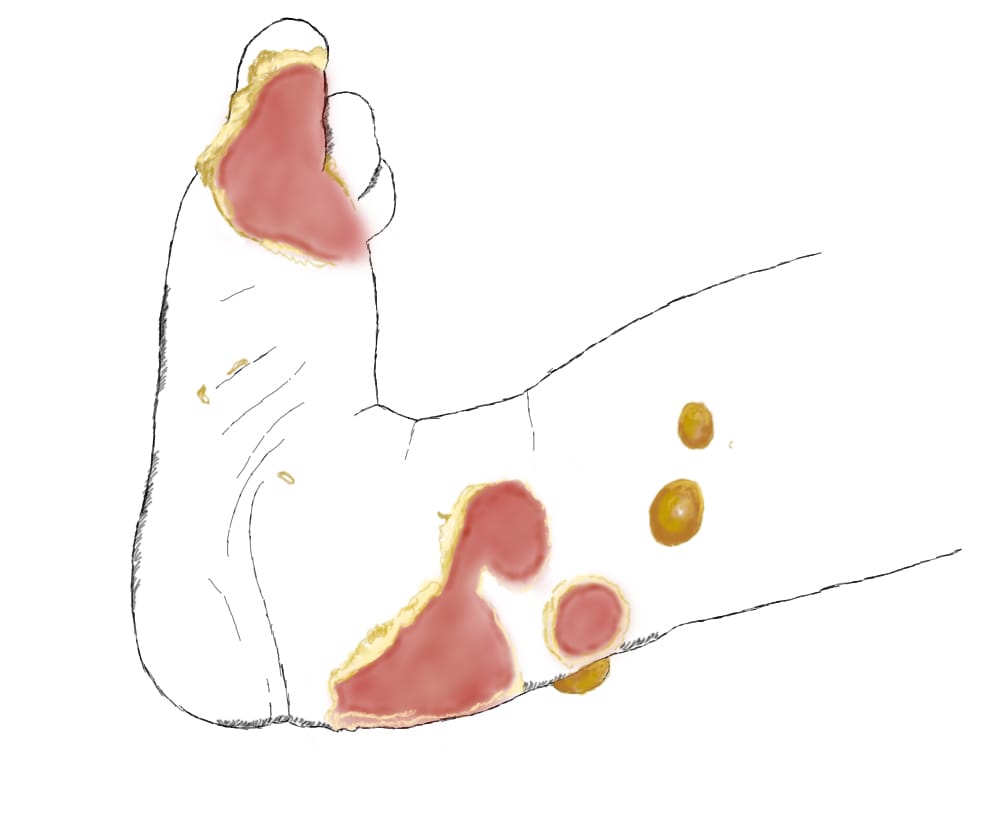
Обширные язвы и эрозии на коже со временем приводят к тому, что она замещается грубой рубцовой тканью. Она отличается плохой чувствительностью, слабой растяжимостью, отсутствием сальных и потовых желёз. Функции кожи утрачиваются, кроме того, рубцы представляют собой и косметический дефект, уродующий внешность.
Рубцовое замещение кожи век приводит к ограничению их движения. Малыш не сможет полноценно открыть или закрыть глаз, вследствие чего орган зрения может остаться без защиты. Изменится и форма глаза вплоть до полного его закрытия. В результате присоединяются конъюнктивиты, блефариты и другие воспалительные заболевания глаза. Ребёнок может и вовсе лишиться зрения.
Рубцы в области рта приводят к формированию микростомии. Ротовая щель стягивается, сужается. В результате малыш не сможет открыть рот до конца. Нарушается процесс глотания и речь.
Грубая рубцовая ткань в области суставов приводит к ограничениям движений в них. Ребёнок не сможет полноценно согнуть и разогнуть сустав, так как рубец не сможет полноценно растянуться, как обычная кожа. Сустав чаще остаётся в одном и том же положении, развивается его контрактура – тугоподвижность.
Эрозии и мокнутия в области пальцев приводят к тому, что рубцовая ткань образуется со срастанием пальцев. В таком случае кисти ребёнка навсегда теряют функцию захвата.
Эрозии на слизистых оболочках также могут заживать с образованием рубцовой ткани, приводя к сужениям – стриктурам пищевода, дыхательных и мочевых путей, кишечника. Ребёнок не может полноценно глотать пищу, говорить, дышать, мочиться, следовательно, будет худеть вплоть до кахексии, часто болеть воспалительными заболеваниями лёгких, почек. Всасывание пищи в кишечнике будет нарушено. Это ещё больше усугубит состояние малыша.
Язвы и эрозии на коже являются входными воротами для разных инфекционных агентов. Поэтому, при несоблюдении правил перевязок, может возникнуть гнойное воспаление кожи, а иногда и системное заражение крови – сепсис.
У людей, страдающих буллёзным эпидермолизом, высок риск развития плоскоклеточного рака кожи.
Симптомы буллезной эмфиземы
В ряде исследований, опубликованных в открытой международной базе PubMed, подчеркивается, что к образованию булл в легких неизбежно приводят такие факторы как:
- Курение (15-20 пачколет);
- Систематическое вдыхание токсических веществ (неблагоприятная экология, работа на вредном производстве, токсикомания, наркомания);
- Низкий уровень качества жизни (скудное и вредное питание, переохлаждение, болезни, отсутствие квалифицированной медицинской помощи).
И только следом за этими факторами по распространенности идут перенесенные в прошлом инфекционно-воспалительные заболевания дыхательных путей, аутоиммунные заболевания, нарушение обмена веществ, врожденные особенности организма.
В преобладающем большинстве случаев буллезную эмфизему диагностируют у людей преклонного или зрелого возраста, однако буллы могут быть обнаружены и у молодых людей.
Небольшие буллы никак себя не проявляют. В целом большинство заболеваний основного дыхательного органа пациенту сложно у себя даже заподозрить — легкие не болят, а когда возникают одышка, кашель, нехватка воздуха, болезнь приводит пациента в реанимацию за считанные часы.








Патологические симптомы могут проявиться по мере увеличения булл и развития эмфиземы. Профилактика заболевания возможна с помощью регулярного скрининга — низкодозовой КТ легких. Обследование в режиме 3D и в высоком разрешении покажет даже малейшие отклонения от нормы.
Для определения (точной стадии) буллезной эмфиземы применяется метод спирометрии — функциональной диагностики легких. Как правило после спирометрии, если показатели не в норме, пульмонолог отправляет пациента на КТ легких.
Заподозрить буллы в легких можно при наличии следующих симптомов:
- Одышка и нарушение дыхания;
- Резистентность к физическим нагрузкам, быстрая утомляемость;
- Гипотония, неконтролируемые аритмии;
- Ощущение постоянной усталости;
- Посинение губ и кончиков пальцев;
- Болезненная бледность;
- Ощущение неполноты вдоха;
- Деформация грудной клетки (становится бочкообразной, если есть крупные буллы).
Поскольку увеличение булл довольно длительно по времени, больной может не замечать ухудшения самочувствия, в частности, становится все труднее задерживать дыхание из-за уменьшения жизненной емкости легких, а гипоксия воздействует в целом на все внутренние органы — человек буквально «увядает».
Причины буллезного эпидермолиза
Основные причины возникновения патологии — мутации более чем в 10 генах, отвечающих за кодирование белков различных слоев кожи, чаще всего в 75% — это гены KRT5 и 14, LAMC2, LAMA3, LAMB3, COL17A1. Как указывает онлайн-ресурс Википедия почти для каждого из установленных подтипов БЭ удалось выявить мутации в определённых генах, среди них чаще всего встречаются:
- миссенс точечные мутации;
- нонсенс точечные мутации;
- делеции и инсерции – потери и соответственно вставки участков хромосом;
- мутации сдвига рамки считывания;
- сплайсинг.
Тип наследования буллезного эпидермолиза встречается как аутосомно-рецессивный (наиболее часто), аутосомно-доминантный, так и однородительская дисомия, и соматический мозаицизм.
Факторы, провоцирующие врожденный буллезный эпидермолиз
Врожденный синдром Бабочки может развиться даже у здоровых родителей, не несущих мутированные гены. Спонтанные мутации возникают внутриутробно и причиной тому становится:
- вредные привычки беременной женщины – курение, злоупотребление алкоголем;
- хаотичный прием лекарственных препаратов и прочие тератогенные факторы.
Почему возникает буллезный эпидермолиз?
Буллезный эпидермолиз вызван дефектами в генах, регулирующих структуру белков, которые формируют кожу и слизистые оболочки, придают им прочность. Т.е. буллезный эпидермолиз – генетическое заболевание. Мутация (повреждение) определенного гена может либо наследоваться ребенком, либо впервые возникать под воздействием различных неблагоприятных факторов. В этом случае мутация в гене возникает во время зачатия или беременности. Причиной повреждений генов может быть радиационное и другие излучения, воздействие вредных токсических и химических веществ, вирусные инфекции и пр.
При наследовании мутации один из родителей ребенка или оба изначально имеет поврежденный ген, при этом он может быть совершенно здоров (носитель). Существует два пути наследования – доминантный и рецессивный. Частота заболеваемости одинакова среди мужчин и женщин.
Форма заболевания зависит от пути наследования (доминантного или рецессивного). Простой буллезный эпидермолиз доминантно наследуется, пограничный – рецессивно, а дистрофические формы – как рецессивно, так и доминантно.
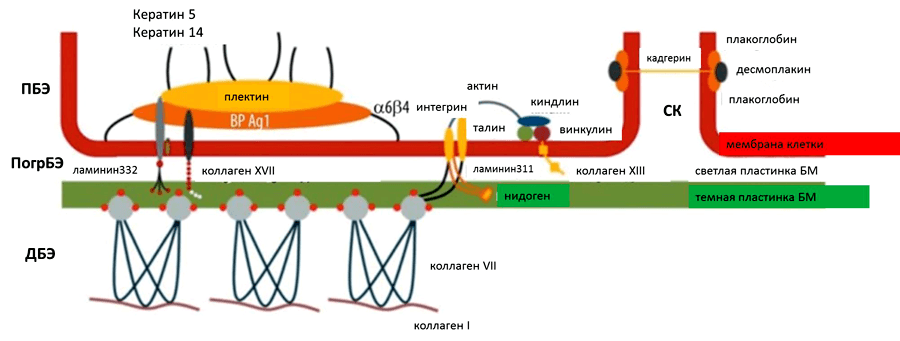
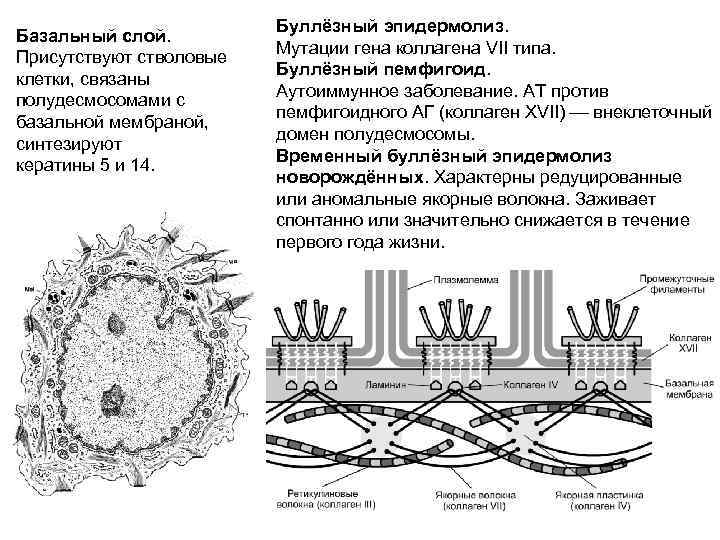
В настоящее время уже известны участки повреждения генов и белки, которые ими кодируются. При простом буллезном эпидермолизе повреждения затрагивают белки кератин 5, кератин 14, плектин, альфа-6,бета-4 интегрин. При пограничной форме заболевания изменения выявляются в белках ламинин 332, коллаген 17 типа, альфа-6,бета-4 интегрин. При дистрофическом буллезном эпидермолизе поражается коллаген 7 типа, при синдроме Киндлера – киндлин – 1. Каждый из этих белков принимает участие в связи верхних и нижних слоев кожи.
Схема «Строение кожи»
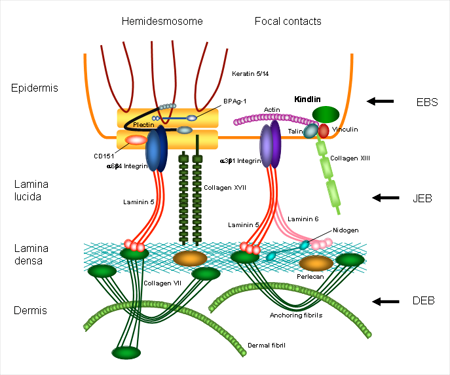
Для формирования более полной картины возникновения заболевания приводим данные о значении, функции этих белков в формировании структуры кожи, а также рассмотреть строение тех уровней кожи, на которых могут происходить патологические изменения.
- Кератин 5 и кератин 14 – участвуют в формировании самого верхнего слоя кожи, осуществляют соединение клеток эпидермиса, поддерживают форму клеток, совместно с коллагеном и эластическими волокнами определяют упругость и прочность кожи.
- Плектин и ламинин 332 – соединяют внутренний скелет клетки с ее оболочкой и просединяет клетки к более глубокому слою кожи.
- Альфа-6,бета-4интегрин – основной рецептор ламинина, т.е. воспринимающая структура.
- Коллаген 17 типа – белок, входящий в комплекс, соединяющий верхние и нижние слои кожи.
- Коллаген 7 типа – главный структурный белок, скрепляющий кожу.
- Киндлин-1 – белок – регулятор активности интегринов.
Все перечисленные белки обеспечивают прочность кожи.
Таким образом, происходит нарушение структуры и функции связывающей слои кожи между собой сложного комплекса клеток, волокон и веществ. И если хотя бы один из компонентов этого комплекса оказывается дефектным, прочность кожи страдает.
Что значит булла в легком?
Согласно определению, данному на международном медицинском CIBA-симпозиуме, буллой следует считать воздушную полость в легком, диаметр которой превышает 1 см, при этом альвеолярная стенка истончается до 1 мм и менее.

В медицине принято разграничивать буллезную эмфизему и буллезную болезнь. Их различают в зависимости от причин возникновения, по этиологии, а также в зависимости от последствий для здоровья. Буллезную эмфизему обычно диагностируют у пациентов с признаками ХОБЛ (хронической обструктивной болезни легких) на фоне двусторонней эмфиземы панацинарного или центрилобулярного типа. При буллезной болезни эмфизема не наблюдается, а полость может быть одна либо немного больше. Буллезная болезнь может быть при врожденном синдроме Марфана. Буллезная эмфизема при отсутствии лечения и адекватных действий по сдерживанию ее роста (отказ от курения, работы на опасном производстве, лечение хронического бронхита и др.) со временем способна привести к дыхательной или сердечной недостаточности, коллапсу легкого, тяжелому кашлю с кровью, к непереносимости физических нагрузок.
Буллы формируются по принципу мыльных пузырей: сначала возникают маленькие, затем они объединяются, образуя крупные буллы, и так далее. При буллезной эмфиземе легочная ткань становится похожа на старую губку — по мере ее изнашивания поры становятся крупнее, а материал теряет эластичность и рвется.
Крупные буллы (диаметр может достигать до 10 см) образуются при разрушении альвеолярных перегородок. Альвеолы — пузырьки, из которых состоит нормальная воздушная легочная ткань. Хранение воздуха, транспортировка кислорода, воздухообмен осуществляется в альвеолах, а сам процесс требует структуры дозированного поступления воздуха. Буллы, даже очень небольшие, привносят элемент хаоса и энтропии в работу важнейшего дыхательного органа.
Диета при буллезном эпидермолизе
Диета 15 стол
- Эффективность: лечебный эффект через 2 недели
- Сроки: постоянно
- Стоимость продуктов: 1600-1800 рублей в неделю
Вне зависимости от возраста больного и типа патологии лечебная диета должна полностью компенсировать потери белков, солей, жидкости, которые вызваны патологическими изменениями внешних покровов и внутренних систем. Если речь идет о детях грудного возраста, то наиболее рекомендованным является материнское молоко, дополненное докормом белковой пищей – не менее 20%. Питание должно быть дробным, механически, термически и химически щадящее, есть следует маленькими порциями. С возрастом в рацион должны быть введены:
- некислые фруктовые соки и пюре;
- нерафинированное растительное масло добавлять в завтрак для облегчения дефекации и насыщения полезными жирами;
- овощи богатые растительной клетчаткой, например, капуста, кабачки, сухофрукты, свекла, которые лучше употреблять перетертыми и отварными.
Под строгий запрет попадает карамель и леденцы, сладости, печенье, спиртные напитки, острые и пряные блюда – любую еду, которая может спровоцировать образование буллезных поражений в пищеводе и других отделах ЖКТ.
Общие сведения
Буллезный эпидермолиз (сокр. БЭ) – это понятие объединяющее целую группу редких наследственных болезней, имеющих генетическую и клиническую гетерогенность. Чаще всего патологические процессы происходят в кератиноцитах и приводят к образованию пузырей с доброкачественным течением, возникновению эрозивных участков кожи и слизистых оболочек, повышенной ранимости и чувствительности кожных покровов к любым незначительным механическим травмам, развитию «механобуллезной болезни», которая еще называется синдром Бабочки. Встречаемость разных видов пузырчатки – всего 1 случай на 30-100 тыс. человек.

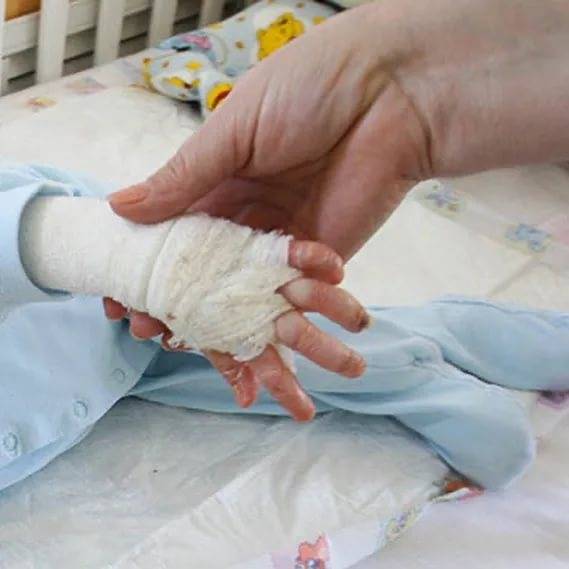
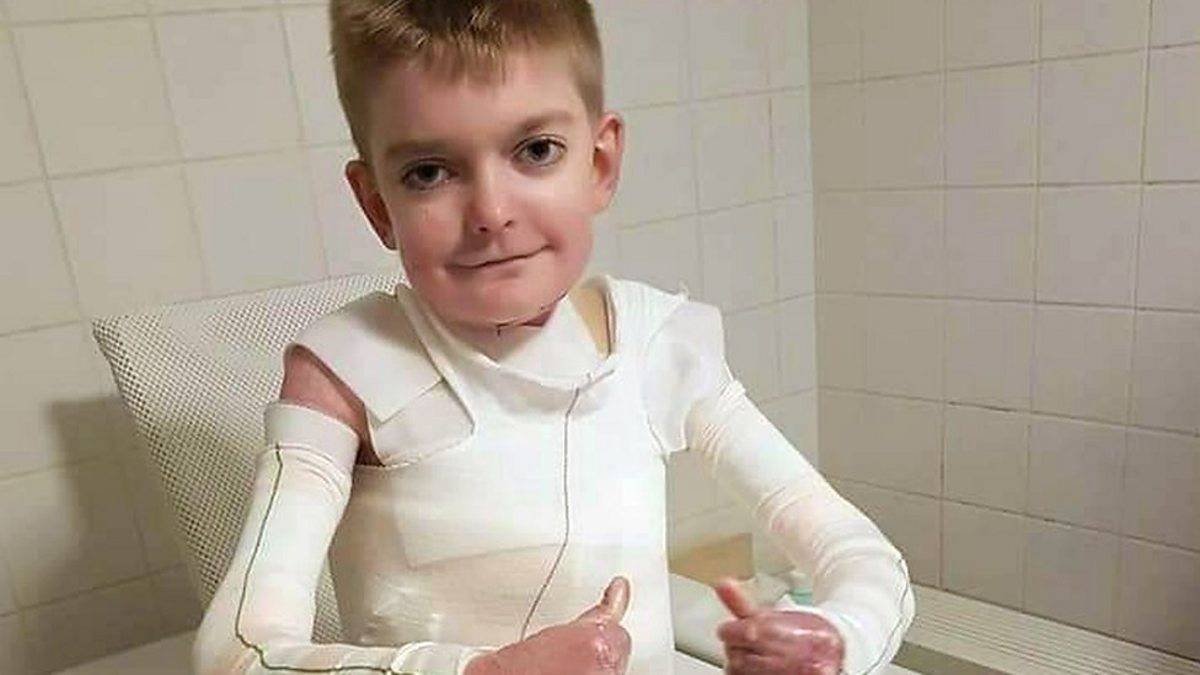
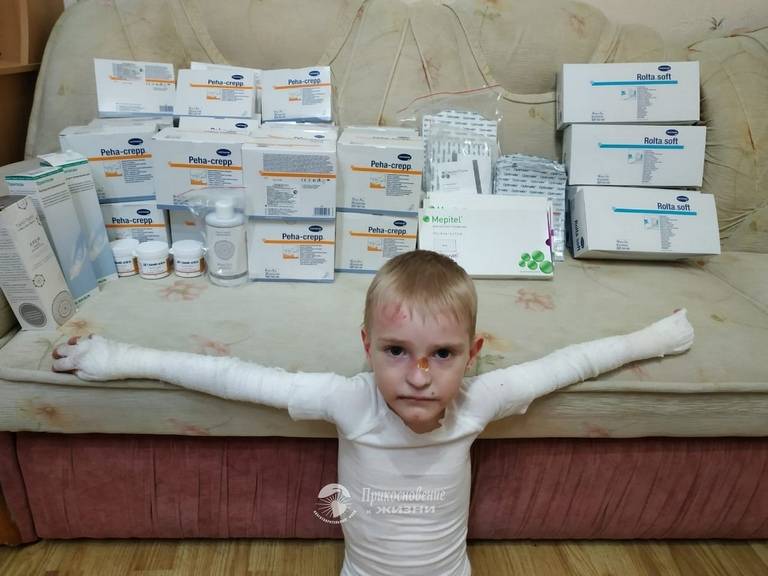
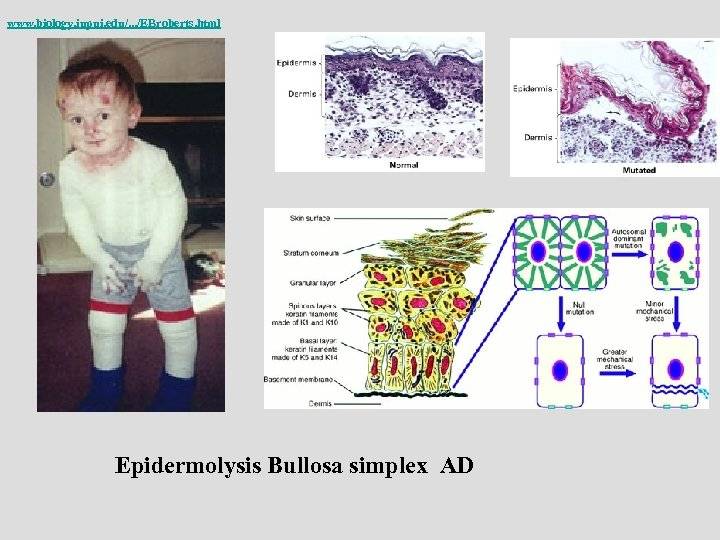


Ввел понятие буллезного эпидермолиза немецкий дерматолог Генрих Кёбнер еще в 1886 г., хотя подобные случаи кожных заболеваний встречались и ранее.
Продолжительность жизни при буллезном эпидермолизе
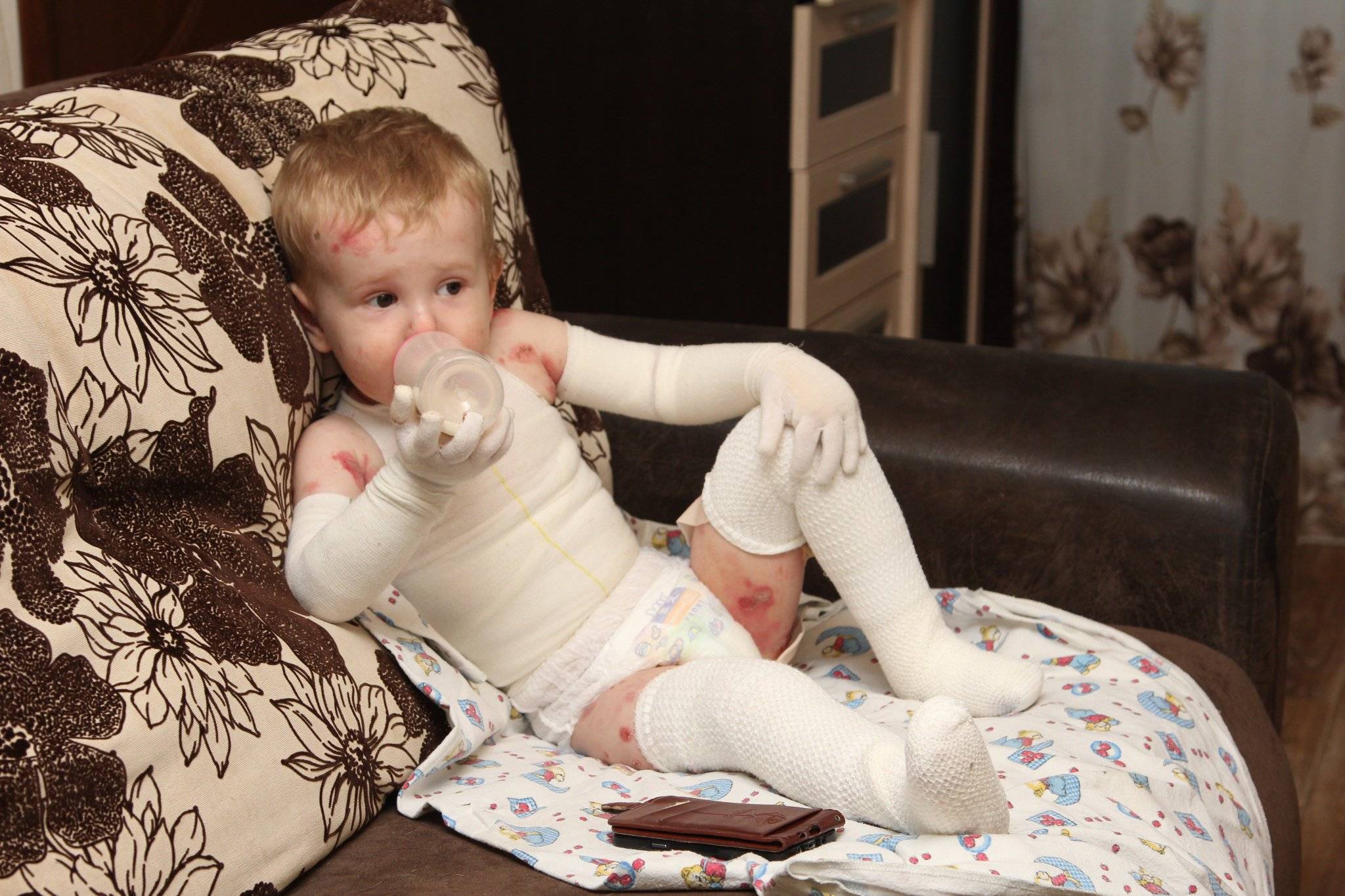
Несмотря на стремительное развитие всемирной медицины продолжительность жизни у пациентов с врожденным эпидермолизом – «детей-бабочек» чаще всего не высокая. Все зависит от подтипа заболевания, глубины и обширности патологических поражений. Простой буллезный эмидермолиз при правильном уходе и комплексной терапии протекает легче, с возрастом минимизируется количество пузырей и прогноз для таких пациентов благоприятный. Другие типы генодерматозной болезни, особенно при присоединении инфекций, чаще всего стафилококковой или стрептококковой, при развитии системных осложнений и сепсиса чаще всего приводят к летальному исходу в ранние годы жизни детей.
Пограничный буллёзный эпидермолиз
При пограничном буллёзном эпидермолизе изменения касаются базальной мембраны эпидермиса. Он подразделяется на генерализованный и локализованный. Общим их признаком является появление пузырей на любом участке тела с обширными поражениями. Также характерны изменения зубной эмали, она истончается, появляются точечные углубления на поверхности зуба. Они часто подвергаются кариесу. Пограничный буллёзный эпидермолиз протекает тяжелее, чем простой.
Генерализованный тяжёлый пограничный буллёзный эпидермолиз ещё называется «летальный». Это смертельное заболевание, исходами которого являются грубое обезображивание и инвалидность. Малыш рождается с пузырями, либо возрастом их проявления служит период новорождённости. Часто пузыри локализуются в периоральной области.
https://youtube.com/watch?v=cC0SyDSdl1M
Ими покрыты волосистая часть головы, ноги, промежность, грудная клетка ребёнка. На кистях и стопах они появляются редко, в отличие от других типов буллёзного эпидермолиза. Исключение составляют конечные фаланги пальцев, на которых располагаются ногти. Сами же пластины разрушаются, отслаиваются и навсегда утрачиваются. Часто высыпаниями поражаются все слизистые оболочки.
Эрозии заживают очень медленно. На их месте кожа атрофируется, образуются грубые рубцы. Из-за возникших осложнений рост малышей останавливается. Они не набирают массу тела. Такие дети часто умирают не прожив и трёх лет от инфекций, истощения и расстройств кровообращения.
Генерализованный средне – тяжёлый пограничный буллёзный эпидермолиз отличается от предыдущего варианта более лёгким течением. Пузыри также могут обнаруживаться у новорождённого ребёнка, но при их заживлении рубцов не образуется. Отличительная черта данного клинического варианта – очаговое выпадение волос и выраженная атрофия кожи волосистой части головы. Растут и развиваются такие малыши в соответствии с возрастом. Болезнь на это никак не влияет.
Лечение
Согласно данным DEBRA существуют следующие направления в лечении буллезного эпидермолиза.
1. Протеиновая терапия – в организм пациента вводится достаточное количество нормального белка. Коррекция клинических проявлений осуществляется путем внутрикожного введения коллагена 7 типа.
2. Генная терапия – вводится ген, кодирующий поврежденный белок. Данный метод осуществляется путем пересадки участка кожи с исправленным геном – трансген (введенный недостатющий ген).
3. Клеточная терапия – вводится необходимое и достаточное количество клеток, содержащих нормальный ген, кодирующий белок. При данном способе лечения в отдельные участки кожи вводят фибробласты здорового донора. Фибробласты синтезируют достаточное количество коллагена для обеспечения соединения слоев кожи (эпидермиса и дермы).
4. Комбинированная терапия – создают «гибридные» кожные трнсплантаты, используя донорские фибробласты и кератиноциты пациента. Используется именно подобное сочетание, т.к. фибробласты с меньшей вероятностью могут вызвать реакцию отторжения трансплантата.
5. Терапия с использованием стволовых клеток костного мозга – в данной области можно выделить два основных направления. Первое, систематическое введение донорских стволовых клеток костного мозга реципиента. Второе, перепрограммирование собственных клеток кожи пациента с целью получения нового источника генетически исправленных стволовых клеток.
6. Лекарственная терапия – включает в себя следующие напрвления:
• «Нокаут и замена» siRNA (информационной РНК)
• Препараты, предотвращающие преждевременное завершение экспрессии генов (PTC124, или гентамицин). Некоторые генетические дефекты при буллезном эпидермолизе заставляют прекратить работать механизм синтеза белка в клетках кожи раньше, чем завершен синтез полноценного белка. Данные препараты препятствуют преждевременному распознаванию стоп-сигнала механизмом синтеза белка, пролонгируя тем самым процесс трансляции.
• Симптоматическая терапия – включает в себя применение анальгетиков, антибиотиков различного спектра действия, противоанемических препаратов, препаратов селена и карнитина (при кардиомиопатии) и пр.
Влияние на качество жизни пациента
Уход за пациентами с БЭ преследует цель контроля заболевания, поскольку до сих пор БЭ не поддается лечению.
Жизни пациентов полны боли и дискомфорта, а стресс постоянно сопровождает их в попытке избежать физического контакта, способного повредить их кожу. Многие дети с БЭ, часто называемые «дети-бабочки», не способны испытывать радости нормального детства. Таких пациентов часто госпитализируют, и люди могут пропускать целые периоды обучения или работы, если они не способны выполнять свои обязанности в результате тяжести повреждений и других последствий заболевания.
БЭ поражает целые семьи. Их жизни во многом подчинены бесконечным процедурам лечения ран и приема медикаментов. Раны требуют ухода: волдыри прокалывают, осушают и перевязывают. Купание и смена повязок могут занимать более трех часов. Следует принимать обезболивающие препараты и антибиотики, посещать врачей, больницы и группы поддержки. Все эти действия создают необходимость ухода за больным БЭ на круглосуточной основе .
Лечение буллезного эпидермолиза
Наиболее эффективным и распространённым на сегодняшний день считается превентивное и симптоматическое лечение, так как радикальной терапии для излечения любого из подтипов БЭ до сих пор разработано не было, хотя точно поставленный диагноз дает возможность существенно повысить качество жизни и снизить вероятность рецидивов.
Наиболее перспективными считаются направления лечения, способные препятствовать образованию или заменять белки кожи неправильной структуры:
- введение белков в организм – протеиновая терапия;
- ведение клеток со здоровыми генами, кодирующими мутированные кожные белки – клеточная терапия;
- генная инженерия, дающая возможность вводить в организм гены, которые заменят имеющиеся мутировавшие;
- использование собственных или донорских стволовых клеток.
Комплексная терапия обычно состоит из таких составляющих как:
- ранозаживляющее лечение с применением Солкосерила, Актовегина, Бепантена, Пантенола и пр.;
- обработка инфицированных участков кожи мазей с антибиотиками, антимикробными составляющими – Дермазином, Левосином, Левомеколем, Фастином, однако более практичным считается изпользование аэрозолей – Олазоля, Гипозоля, Легразоля, Левовинизоля и т.д.;
- трансплантация бионической кожи или донорской от близких родственников;
- использование препаратов, ингибирующих синтез или активность коллагеназы, в том числе ретиноидов, Фенитоина, Эритромицина, высоких доз витамина Е;
- в целях предупреждения риска инфицирования проводят облучение пораженных участков кожи при помощи ультрафиолетовых ламп в субэритемных дозах;
- сведение до минимума травматизации кожных покровов – использования одежды и пеленок без швов, резинок, сделанных из натуральных гипоаллергенных мягких тканей;
- назначение препаратов – антибиотических, противогрибковых и пр. при выявлении присоединении вторичной инфекции;
- полоскание рта отварами ромашки, календулы, зверобоя, шалфея, коры дуба, корневищ змеевика;
- при выраженной анемии проводят переливание эритроцитарной массы, альбуминов, плазмы;
- общеукрепляющая терапия может быть дополнена анаболическими средствами, поливитаминными комплексами, диетой;
- может быть назначена поддерживающая психотерапия и антидепрессанты.
Доктора
специализация: Генетик / Дерматолог

Головинов Андрей Иванович
10 отзывовЗаписаться
Подобрать врача и записаться на прием
Лекарства
Актовегин
Бепантен
Пантенол
Дермазин
Левосин
Левомеколь
Олазоль
Фенитоин
Эритромицин
Преднизолон
Радевит
Наиболее эффективными на сегодняшний день являются препараты, способные блокировать процессы кодирования белков с нарушенной структурой.
Кроме того, в тяжелых случаях назначают антигистаминные препараты, а также:
- при сильном зуде могут быть назначены седативные средства;
- благодаря глюкокортикоидам, к примеру, Преднизолона удается снять воспаление и неприятные ощущения;
- для купирования болевого синдрома назначают анальгетики;
- использование противошоковых препаратов;
- пациент может быть введен в индуцированную медикаментозную кому.
Процедуры и операции
При механобуллезном эпидермолизе очень важны гигиенические процедуры по обработке пузырей, ран, язвенных и эрозивных изменений кожи, так как это повышает вероятность и скорость заживления и зарастания, не дает распространяться очагу поражения. Для этого следует использовать из особого материала не прилипающие повязки и бинты, у которых не вылазят волокна и не травмируют тонкую нежную кожу «человека-бабочки». Сначала рекомендовано произвести асептическое вскрытие пузырей или их опорожнение, затем обработать их крышки антисептическим средством и наложить слой гелиомициновой мази, Коласпона, Альгикола либо Дигиспона.
Кожные покровы нужно ежедневно ожиривать – наносить негустые мази и кремы на водно-эмульсионной основе, содержащие ретинол с пальмитатом, растительные масла, мочевину, к примеру, Радевит.
Наиболее приятными процедурами являются ванны с травами, обладающими вяжущим и противовоспалительным действием.
Как передается заболевание?
Все формы простого буллезного эпидермолиза наследуются доминантно. Это значит, что если один из родителей болен, он может передать заболевание своим детям. Вероятность заболеть у ребенка 50 %. Такая вероятность существует при каждой беременности, независимо от того, есть ли уже больной ребенок в семье или нет. Ситуация также не меняется в зависимости от того, болен ли отец или мать (то есть нет зависимости от пола). Заболевание встречается одинаково часто у женщин и мужчин. Через поколение заболевание передать невозможно.


Нередко встречаются случаи, когда ни у кого из родственников буллезного эпидермолиза нет. Появление в семье больного ребенка объясняется «первичной мутацией» в результате повреждения гена во время зачатия, до него или после. Причиной повреждений может быть повышенная радиация (облучение), воздействие токсических веществ, тяжелые инфекционные заболевания. Буллезный эпидермолиз, развившийся в результате первичной мутации, передается следующему поколению с той же вероятностью, что и наследственный.
Форма заболевания никогда не изменяется в течение жизни, простой БЭ не может превратиться в дистрофический. Если диагноз через несколько лет изменен, то это можно отнести за счет не совсем четкой симптоматики в раннем возрасте, и в результате – ошибки в определении формы заболевания. Больной простым БЭ не может иметь ребенка, больного дистрофическим БЭ. Более того, проявления заболевания и его течение в одной и той же семье очень сходны.
Анализы и диагностика
Наибольшее значение в постановке диагноза играет биопсия — исследование образцов кожи при помощи трансмиссионных электронных микроскопов, позволяющих осуществить визуализацию и провести полуколичественный анализ различных структур эпидермиса. Благодаря доступности моноклональных и поликлональных антител против белков разных слоев эпидермиса, участвующих в патогенезе буллезного эпидермолиза на сегодняшний день всю большую популярность завоёвывают иммуногистологические методы.
Иммуногистохимический метод исследования и метод непрямой иммунофлюорисценции позволяют определить состояние экспрессии структурных белков с наследственными дефектами в клетках кожи — кератиноцитах и базальных мембранах (основы эпителия, выполняющей барьерную и трофическую функцию), а также схему распределения протеидов в ранее образованных или искусственно спровоцированных пузырях, в том числе — их глубину локализации.
Благодаря современным методам ДНК-диагностики удается быстро классифицировать разновидность патологии, выявить структурные белки, подвергшиеся мутации и составить клинический прогноз. Инновационный метод генетического анализа – прямое секвенирование дает возможность выявить мутации, их тип и локализацию, достоверно подтвердить диагноз.
Помимо этого важную роль играет сбор семейного анамнеза и истории болезни пациента, комплексное обследование всего организма, проведение лабораторных анализов.