Медикаментозное лечение эпилепсии.
История
лечения эпилепсии начинается с появления солей брома, которые первым применил
Чарльз Лекок в 1857 г.
С 1912 г.
единственными препаратами для купирования эпилептических припадков стали
барбитураты. Они были созданы и внедрены в клиническую практику профессором
органической химии Берлинской промышленной Академии Адольфом фон Байером в 1863 г. и названы так в
честь его жены Барбары. В 1904
г. Эмиль Фишер и Иосиф Меринг открыли соли барбитуровой
кислоты (барбитал и фенобарбитал). Барбитал в дальнейшем получил название
«веронал» в честь города Верона (Северная Италия), где разыгралась трагедия
Ромео и Джульетты — так назывался сонный напиток, который монах дал Джульетте.
Начиная с 1938 г.,
после клинической апробации гидантоинов (фенитоин, дилантин, дифенин), а затем
и других противоэпилептических средств (тримидон, триметатион, триметин,
1946г.), примидона (гексамидин, 1952г.), этосуксимида (суксилеп, 1960 г.) появились новые
возможности воздействия на эпилептические припадки. С этого времени наступил
этап полипрагмазии в лечении эпилепсии, при котором приоритетной стала тактика
назначения одновременно нескольких противоэпилептических препаратов (метод
«шрапнельного выстрела»). Тенденция к полипрагмазии привела к использованию
различных комбинаций противоэпилептических препаратов. Чаще всего комбинировали
барбитураты, подавляющие эпилептическую активность на корковом уровне с
гидантоинами, действующими на уровне стволовых образований мозга, добавляя к
ним стимулирующее вещество с целью уменьшения седативного действия (кофеин,
эфедрин, фенамин, стрихнин). Такие лекарственные смеси носили наименования по
фамилиям авторов, их предложивших (Серейского, Воробьева, Расина, Кармановой,
Бродского). Полипрагмазия как основной метод лечения эпилепсии применялась
вплоть до 60–70-х годов ХХ в., т.к. считалось необходимым использовать
одновременно несколько противоэпилептических средств (Ремезова Е.С., 1954;
Серейский М.Я., 1955; Воробьев С.П., 1958; Ходос Х.Д., 1964; Абрамович Г.Б.,
Харитонов Р.А., Тец И.С., 1965; Вайнтруб М.Я., 1979 и другие).
В 1964 г. в клиническую
практику был введен карбамазепин (финлепсин, тегретол), а в 1967 г. — вальпроаты
(депакин, конвулекс, конвульсофин). Это постепенно изменило представление о
преимущественном значении комбинированной терапии для лечения эпилепсии. В
результате «золотым стандартом» в лечении эпилепсии в конце 80-х годов прошлого
столетия стала монотерапия (использование одного препарата), а политерапия
(применение двух и более антиконвульсантов) стала применяться лишь при
невозможности адекватной монотерапии.
Стратегия лечения эпилепсии
1. Установление диагноза эпилепсии
(анатомо-электро-клинически, т.е. с обязательным использованием, кроме
клинического исследования, результатов электроэнцефалограммы и
магнитно-резонансной томографии).
2. Выбор АЭП в соответствии с формой
эпилепсии и типом припадков.
3. Начало лечения с препарата с наиболее
широким терапевтическим диапазоном и минимальной вероятностью осложнений.
4. Доза АЭП должна быть не ниже
рекомендуемой терапевтической из расчета мг/кг веса в сут или мг/сут для данного
возраста.
5. Начало с монотерапии с доведением дозы
до эффективной. Лишь при максимально допустимой дозе при отсутствии
положительного эффекта — переход на АЭП второго выбора, желательно последнего
поколения (альтернативная монотерапия).
6. При отсутствии эффекта от
альтернативной монотерапии — рациональная политерапия (комбинация двух, в
крайнем случае, трех АЭП) с обязательным включением АЭП последнего поколения.
7. Регулярность приема АЭП.
8. Продолжительность приема АЭП в
зависимости от формы эпилепсии и эффективности лечения.
Выбор
антиэпилептического препарата (АЭП) в зависимости от типа эпилептического
припадка (в порядке убывания)
Тип | Препараты выбора |
Парциальные (простые, сенсорные, с нарушением | 1–4. Карбамазепин, окскарбазепин, топирамат, 5–6. Вальпроаты, ламотриджин |
Генерализованные абсансы (типичные, атипичные) | 1–2. Вальпроаты, ламотриджин. 3–4. |
Тонико-клонические, тонические, клонические | 1–6. |
Миоклонические | 1. 2. 3. 4. |
Атонические | 1. 2–3. 4. |
Вегетативно-висцеральные | 1. 2–3. 4. |














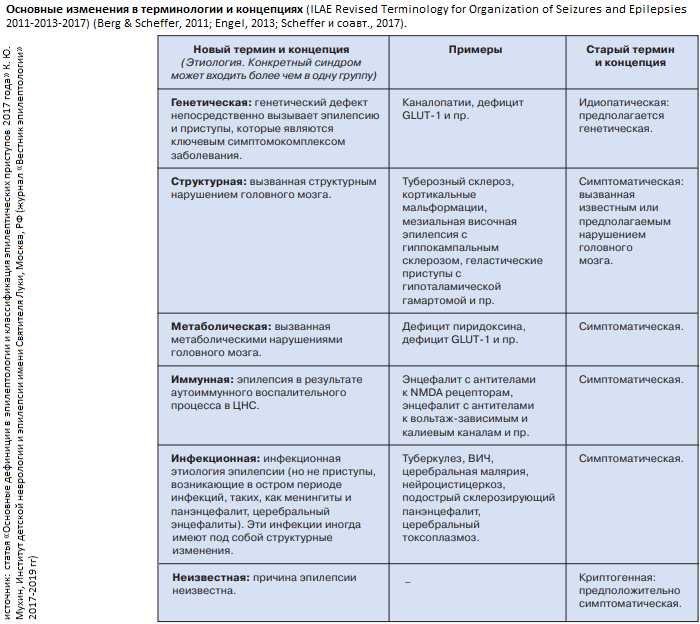





Выделяют следующие виды приступов:
- Генерализированные судорожные припадки заключаются в резком напряжении мышц и кратковременной остановкой дыхания. Далее начинаются судороги, продолжительностью от 10 секунд до 20-ти минут. Приступ прекращается самостоятельно, после него ребенок засыпает.
- Безсудорожные генерализированные приступы (абсансы) менее выражены. Они заключаются во внезапных замираниях, взгляд больного становится отсутствующим. Иногда у ребенка могут подрагивать веки, закрываться глаза. Длительность – около 5-20-ти секунд, в остром периоде ребенок не реагирует на внешние раздражители. По окончанию он может дальше заниматься своими делами. Обычно детская эпилепсия этого типа начинается в возрасте 5-7 лет и чаще проявляется у девочек. После полового созревания ребенок может «перерасти» болезнь. Но не стоит этого ждать, так как абсансная форма может перейти в более сложную.
- Атонические приступы характеризуются внезапными потерями сознания с расслаблением мышц. Их легко спутать с обмороками. Если приступы случаются часто – это серьезный повод обратиться к неврологу и детскому эпилептологу.
- Детский спазм – это непроизвольные наклоны головы или туловища вперед с прижатием рук к груди. Приступы могут случаться после пробуждения, особенно резкого, и свойственны детям 3-х лет. К пяти годам детская эпилепсия этого типа может или пройти совсем или же перейти в другую форму.
Кроме описанных типов существуют и другие формы. Если в состоянии ребенка вас что-то настораживает – стоит обратиться за консультацией к детскому эпилептологу.
Признаки и симптомы
Под термином «эпилепсия» скрыто более 60-ти патологий нервной системы. Существуют несколько характерных признаков, при которых стоит обратиться к эпилептологу как можно скорее:
- Резкие интенсивные головные боли, сопровождающиеся рвотой, спутанностью сознания, нарушениями речи;
- Внезапные судороги длительностью от нескольких секунд до 10-20 минут;
- Потеря сознания;
- Резкое напряжение или расслабление мышц, сопровождающееся остановкой дыхания, непроизвольным мочеиспусканием;
- Замирания в необычных положениях, со странной мимикой и/или полным отсутствием реакций на внешние раздражители.
Эпилепсия у детей (особенно новорожденных) может протекать со смазанными симптомами, что вызывает сложности при диагностике.
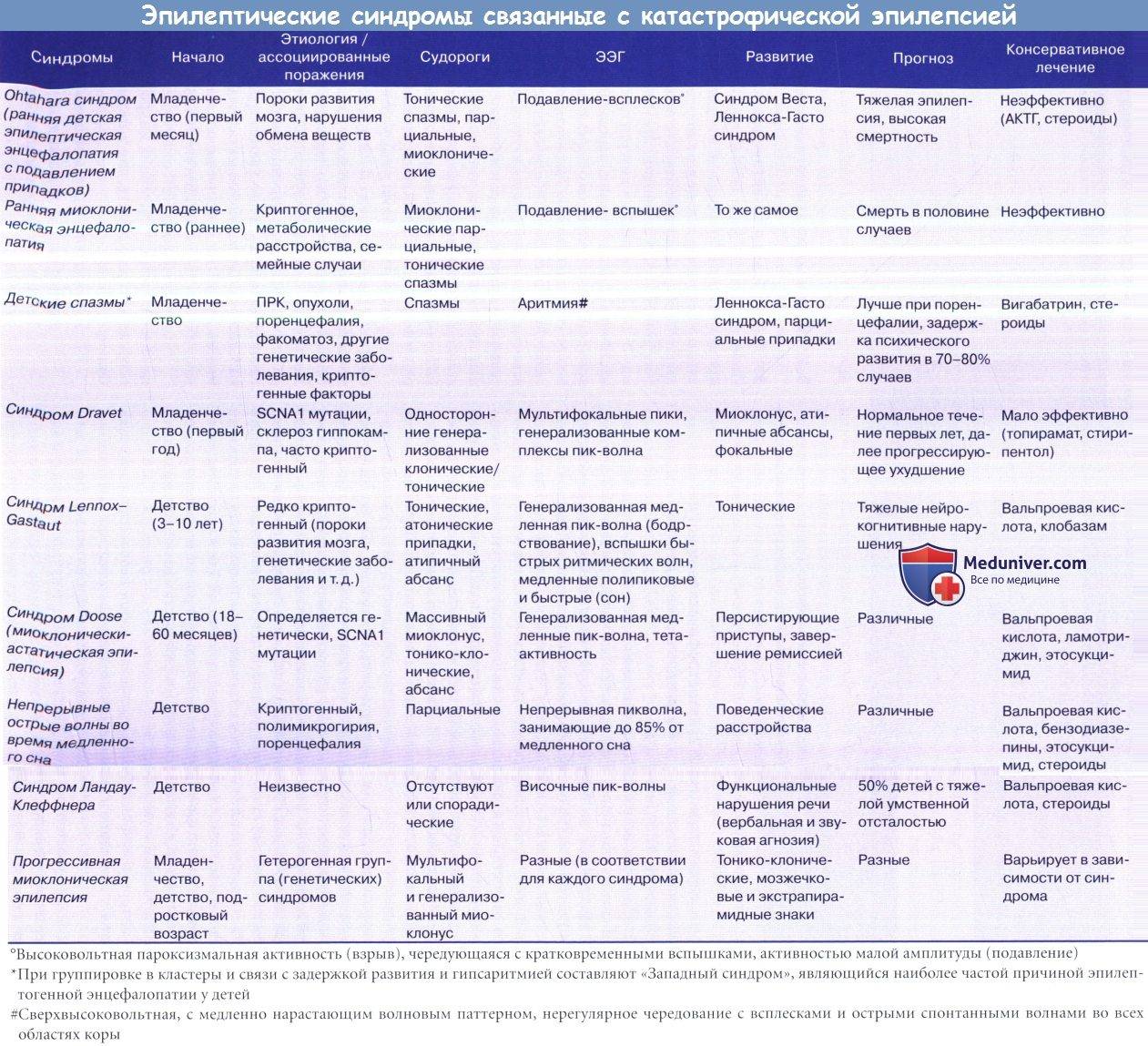
Синдром Леннокса-Гасто.
Синдром Леннокса-Гасто (СЛГ) – эпилептическая энцефалопатия детского возраста, характеризующаяся полиморфизмом приступов, специфическими изменениями ЭЭГ и резистентностью к терапии. Частота СЛГ составляет 3-5% среди всех эпилептических синдромов у детей и подростков; болеют чаще мальчики.
Заболевание дебютирует, преимущественно, в возрасте 2-8 лет (чаще 4-6 лет). Если СЛГ развивается при трансформации из синдрома Веста, то возможно 2 варианта:
Инфантильные спазмы трансформируются в тонические приступы при отсутствии латентного периода и плавно переходят в СЛГ.
Инфантильные спазмы исчезают; психомоторное развитие ребенка несколько улучшается; картина ЭЭГ постепенно нормализуется. Затем спустя некоторый латентный промежуток времени, который варьирует у разных больных, появляются приступы внезапных падений, атипичные абсансы и нарастает диффузная медленная пик-волновая активность на ЭЭГ.
Для СЛГ характерна триада приступов: пароксизмы падений (атонически- и миоклонически-астатические); тонические приступы и атипичные абсансы. Наиболее типичны приступы внезапных падений, обусловленные тоническими, миоклоническими или атоническими (негативный миоклонус) пароксизмами. Сознание может быть сохранено или выключается кратковременно. После падения не наблюдается судорог, и ребенок сразу же встает. Частые приступы падений приводят к тяжелой травматизации и инвалидизации больных.
Тонические приступы бывают аксиальными, проксимальными или тотальными; симметричными либо четко латерализованными. Приступы включают в себя внезапное сгибание шеи и туловища, подъем рук в состоянии полуфлексии или разгибания, разгибание ног, сокращение лицевой мускулатуры, вращательные движения глазных яблок, апноэ, гиперемию лица. Они могут возникать, как в дневное время, так и особенно часто, ночью.
Атипичные абсансы также характерны для СЛГ. Проявления их многообразны. Нарушение сознания бывает неполным. Может сохраняться некоторая степень двигательной и речевой активности. Наблюдается гипомимия, слюнотечение; миоклонии век, рта; атонические феномены (голова падает на грудь, рот приоткрыт). Атипичные абсансы обычно сопровождаются понижением мышечного тонуса, что вызывает как бы “обмякание” тела, начиная с мышц лица и шеи.
В неврологическом статусе отмечаются проявления пирамидной недостаточности, координаторные нарушения. Характерно снижение интеллекта, не достигающее, однако, тяжелой степени. Интеллектуальный дефицит констатируется с раннего возраста, предшествуя заболеванию (симптоматические формы) или развивается сразу после появления приступов (криптогенные формы).
При ЭЭГ-исследовании в большом проценте случаев выявляется нерегулярная диффузная, часто с амплитудной асимметрией, медленная пик-волновая активность с частотой 1,5-2,5 Гц в период бодрствования и быстрые ритмические разряды с частотой около 10 Гц – во время сна.
При нейровизуализации могут иметь место различные структурные нарушения в коре головного мозга, включая пороки развития: гипоплазия мозолистого тела, гемимегалэнцефалия, кортикальные дисплазии и пр.
В лечении СЛГ следует избегать препаратов, подавляющих когнитивные функции (барбитураты). Наиболее часто при СЛГ применяются вальпроаты, карбамазепин, бензодиазепины и ламиктал. Лечение начинается с производных вальпроевой кислоты, постепенно увеличивая их до максимально переносимой дозы (70-100 мг/кг/сут и выше). Карбамазепин эффективен при тонических приступах – 15-30 мг/кг/сут, но может учащать абсансы и миоклонические пароксизмы. Ряд больных реагирует на увеличение дозы карбамазепина парадоксальным учащением приступов. Бензодиазепины оказывают эффект при всех типах приступов, однако этот эффект временный. В группе бензодиазепинов применяются клоназепам, клобазам (фризиум) и нитразепам (радедорм). При атипичных абсансах может быть эффективен суксилеп (но не как монотерапия). Показана высокая эффективность комбинации вальпроатов с ламикталом (2-5 мг/кг/сут и выше). В США широко используется комбинация вальпроатов с фелбаматом (талокса).
Прогноз при СЛГ тяжелый. Стойкий контроль над приступами достигается лишь у 10-20% больных. Прогностически благоприятно преобладание миоклонических приступов и отсутствие грубых структурных изменений в мозге; негативные факторы – доминирование тонических приступов и грубый интеллектуальный дефицит.
Юношеская абсанс эпилепсия.
Юношеская абсанс эпилепсия (ЮАЭ) – разновидность идиопатической генерализованной эпилепсии, которая характеризуется основным видом приступов – абсансами, дебютирующими в пубертатном периоде с высокой вероятностью присоединения ГСП и характерными ЭЭГ-изменениями в виде генерализованной пик-волновой активности с частотой 3 Гц и более.
Дебют абсансов при ЮАЭ варьирует от 9 до 21 года, в среднем, 12.5 лет. У значительного большинства больных (75%) абсансы начинаются в сравнительно коротком временном промежутке – 9-13 лет
Важной особенностью ЮАЭ является частый дебют заболевания с ГСП – 40% случаев.
Абсансы у пациентов, страдающих ЮАЭ, проявляются коротким выключением сознания с застыванием и гипомимией. Характерно значительное преобладание простых абсансов, то есть приступов без какого – либо двигательного компонента
Продолжительность приступов составляет от 2 до 30 сек, в среднем, 5-7 сек. Вместе с тем, у половины пациентов отмечаются очень короткие абсансы, не превышающие 3 сек. Характерной особенностью ЮАЭ является и относительно невысокая частота приступов по сравнению с ДАЭ. У большинства больных преобладают единичные абсансы в течение дня или 1 приступ в 2-3 дня.
Генерализованные судорожные приступы констатируются у большинства больных – 75%. В группе больных с ГСП заболевание чаще дебютирует не с абсансов, а с тонико-клонических судорожных пароксизмов. ГСП характеризуются короткими нечастыми тонико-клоническими судорогами, возникающими, обычно, при пробуждении или засыпании.
В отличие от ДАЭ, гипервентиляция провоцирует возникновения абсансов не более чем у 10% больных ЮАЭ. ГСП у 20% больных провоцируются депривацией сна.
При ЭЭГ-исследовании в межприступном периоде результаты, близкие к норме, констатируются у 25% пациентов. Основным ЭЭГ-паттерном является генерализованная пик-волновая активность с частотой 3 Гц и более (4-5 в сек), носящая, преимущественно, симметричный и билатерально- синхронный характер.
Лечение. Эффективность лечения при ЮАЭ достоверно ниже, чем при ДАЭ. Терапевтическая ремиссия достигается, в среднем, у 60% больных, значительное урежение приступов – 35%, отсутствие эффекта – 5%. Лечение начинается с монотерапии препаратами вальпроевой кислоты. Средняя дозировка составляет 30-50 мг/кг/сут. Ввиду крайне высокой вероятности при ЮАЭ присоединения ГСП, начинать лечение с сукцинимидов, а также применять их в виде монотерапии, категорически противопоказано. При отсутствии существенного эффекта от монотерапии вальпроатами в достаточно высоких дозах, применяется комбинация вальпроатов с сукцинимидами или ламикталом. Средние дозы суксилепа составляют 20 мг/кг/сут.; ламиктала – 1-5 мг/кг/сут.
Признаки патологии
У височной эпилепсии признаки проявляются по-разному. Это зависит от области, в которой расположен очаг патологии. Специалисты выделяют три вида приступов: простые, сложные, вторичные.
Простые фокальные припадки проявляются ощущением дежавю. Постепенно ребенок перестает узнавать родителей и предметы. Он может слышать, ощущать запахи и прикасаться к тому, что не является реальным. Возникают панические атаки, беспричинный страх или наоборот — внезапное чувство счастья, восторга.
Сложные эпилептические припадки проявляются в виде изменений в поведении и сознании — неконтролируемые движения рта, конечностей и странные слова. При височной эпилепсии приступы продолжаются не более 2 минут.
Вторичные генерализованные тонико-клонические припадки вначале проявляются в форме простых или сложных парциальных приступов. При височной эпилепсии симптомы усиливаются: появляются подергивания и судороги, по мере того как болезнь распространяется, постепенно охватывая весь головной мозг.
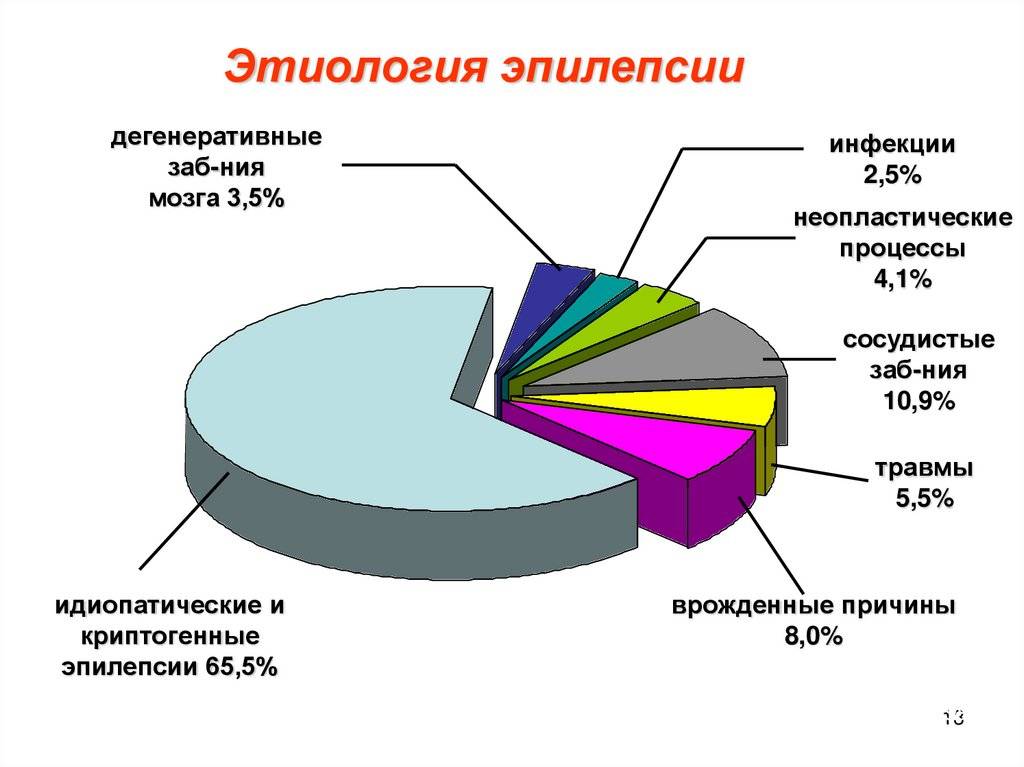
Причины развития заболевания
Эпилепсию височной доли определяют по эпилептическим приступам, спровоцированным сосудистыми патологиями и новообразованиями в головном мозге. Риск развития заболевания повышается при родовых травмах, ушибах головы и герпетической инфекции. У височной эпилепсии причина зачастую кроется в генетической предрасположенности.
Существуют этиологические факторы, способствующие развитию заболевания:
- черепно-мозговые травмы в анамнезе;
- инфекционные заболевания ЦНС: бруцеллез, поствакцинальный энцефаломиелит, вирус герпеса, клещевой энцефалит, сифилитическое поражение головного мозга, японский комариный энцефалит, гнойный менингит;
- ишемический или геморрагический инсульт.
Спровоцировать развитие эпилепсии способны другие патологии — ограниченное гнойное расплавление вещества мозга, внутримозговое кровоизлияние, туберозный склероз (болезнь Бурневилля), аневризма сосудов головного мозга, различные опухолевые образования — астроцитомы, ангиомы, глиомы, гломусные новообразования.
Юношеская миоклоническая эпилепсия.
Юношеская миоклоническая эпилепсия (ЮМЭ) – одна из форм идиопатической генерализованной эпилепсии, характеризующаяся дебютом в подростковом возрасте с возникновением массивных билатеральных миоклонических приступов, преимущественно в руках, в период после пробуждения пациентов. ЮМЭ – одна из первых форм эпилепсии с известным генетическим дефектом. Предполагается двулокусная модель наследования (доминантно-рецессивная), причем доминантный ген локализован на коротком плече хромосомы 6.
Дебют ЮМЭ варьирует от 7 до 21 года с максимумом в возрастном интервале 11-15 лет. Заболевание может начаться в более раннем возрасте с абсансов или ГСП, с последующим присоединением миоклонических приступов в пубертатном периоде. Миоклонические приступы характеризуются молниеносными подергиваниями различных групп мышц; они чаще двухсторонние, симметричные, единичные или множественные, меняющиеся по амплитуде. Локализуются, главным образом, в плечевом поясе и руках, преимущественно в разгибательных группах мышц. Во время приступов больные роняют предметы из рук или отбрасывают их далеко в сторону. У 40 % пациентов миоклонические приступы захватывают и мышцы ног, при этом больной ощущает как бы внезапный удар под колени и слегка приседает или падает (миоклонически-астатические приступы); затем тут же встает. Сознание во время приступов обычно сохранено. Миоклонические приступы возникают или учащаются в утренние часы, после пробуждения пациента. В 90 % случаев они сочетаются с ГСП пробуждения и в 40 % – с абсансами. Основными провоцирующими приступы факторами являются депривация сна и внезапное насильственное пробуждение. Примерно 1/3 больных ЮМЭ (чаще женского пола) обнаруживают фотосенситивность.
Эпилептическая активность на ЭЭГ выявляется у 85% больных в межприступном периоде. Наиболее типична генерализованная быстрая (от 4-х Гц и выше) полипик-волновая активность в виде коротких вспышек. Возможно также появление пик-волновой активности 3 Гц.
Лечение. Наряду с медикаментозной терапией необходимо строго придерживаться соблюдения режима сна и бодрствования; избегать недосыпания и факторов фотостимуляции в быту. Базовые препараты – исключительно производные вальпроевой кислоты. Средняя суточная дозировка – 40-60 мг/кг. При недостаточной эффективности назначается политерапия: депакин + суксилеп (при резистентных абсансах); депакин + фенобарбитал или гексамидин (при резистентных ГСП); депакин + ламиктал или клоназепам (при резистентных миоклонических приступах и выраженной фотосенситивности).
Полная медикаментозная ремиссия достигается у 75% больных, причем в большинстве случаев на монотерапии вальпроатами. Однако в последующем при отмене АЭП, рецидивы констатируются у половины больных. При данной форме эпилепсии рекомендуется отмена АЭП спустя не менее 4-х лет с момента наступления ремиссии.






Причины судорог у собак и кошек
Одна из таких причин — гипоксия или аноксия (недостаточное снабжение или временное отсутствие притока кислорода к головному мозгу), вызванная нарушением дыхания или сердечной деятельностью. Судороги могут быть связаны с пониженным содержанием в крови кальция (гипокальцемия) или глюкозы (гипогликемия), болезнями почек, заболеваниями печени, диабетом, а также послеродовой титанией (заболевание, возникающее в период лактации после родов).
Среди других причин, способных вызвать сокращения в мышцах у кошек или собак, можно выделить паразитарные заболевания, тепловой удар, заболевания щитовидной железы, интоксикацию (отравление растениями или попаданием в организм ядохимикатов).Довольно часто ветеринарные врачи сталкиваются с отравлениями теми средствами, которые предназначены для уничтожения насекомых и грызунов. Очень опасным ядом считается мышьяк. Также судороги могут быть спровоцированы гидроцефалией (водянкой головного мозга), черепно-мозговой травмой, опухолью мозга, сосудистыми заболеваниями, а также хроническими заболеваниями центральной нервной системы, которые сопровождаются поражением головного мозга. К патологиям головного мозга, способным вызвать судороги у собаки, кошки или другого домашнего животного, кроме истинной эпилепсии, относятся, к примеру, воспалительные заболевания. Чаще всего они представляют собой осложнения при разных инфекциях (тосксоплазмоз, чума, бешенство и другие), бактериальных поражениях мозга.
В зависимости от того, какое именно заболевание стало причиной появления недуга, будет меняться и его характер. По своему типу судороги делятся на несколько видов и имеют некоторые различия.
Диагностика заболевания
Эпилепсия височных долей диагностируется с применением высокотехнологичных методик, способных выявить самые серьезные патологии. Обычно назначают следующие обследования:
- анализы крови и мочи;
- электроэнцефалографию;
- КТ и МРТ — помогают обнаружить изменения структуры головного мозга, новообразования, сосудистые аномалии, атрофию тканей в лобно-височной доле;
- полисомнографию с пробой депривации — обнаруживает патологическую активность отделов головного мозга в состоянии сна.
У детей эпилепсия диагностируется своевременно только благодаря участию родителей. Они первые обнаруживают у своих детей отклонения в поведении и самочувствии, связанные с «отключениями» сознания, и незамедлительно обращаются к врачу.