Диагностика
Обычно выявление синдрома не составляет труда из-за характерной клинической картины недуга и состоит из несколько этапов:
определение внешних признаков.
При обследовании пациента обращает на себя внимание характерный внешний вид, отставание в умственном и физическом развитии. Нередко врач замечает хрипловатый, сиплый голос у ребёнка, мышечную слабость, косоглазие, пупочную и паховые грыжи;. лабораторные исследования
лабораторные исследования.
Исследуя кровь биохимическими методами можно обнаружить повышенную концентрацию кальция, холестерина, тиреотропного гормона.
Благодаря молекулярно-генетическому анализу определяется мутация в 7 хромосоме, что помогает удостовериться в генетической природе заболевания, определить выраженность патологии;

инструментальные методы.
С помощью своевременного комплексного обследования можно выявить признаки нарушений в различных органах, вовремя назначить соответствующее лечение. Используя такие методы, как ЭхоКС, ЭКГ определяются патологии органов кровообращения, нарушения сердечного ритма. Ультразвуковое исследование органов брюшной полости необходимо для исключения дисфункций в работе внутренних органов, в первую очередь почек.
Рентгенологическое исследование поможет определить и подобрать правильное лечение при заболеваниях опорно-двигательного аппарата (деформациях позвоночника, грудины, стопы). С помощью магнитно-резонансной томографии можно определить признаки церебральной гипоплазии, уменьшение задней части мозолистого тела и объёма белого вещества головного мозга;
консультации специалистов.
Для подтверждения патологий малыша консультируют кардиолог, офтальмолог, травматолог, ортопед, стоматолог, невропатолог и другие специалисты.
Профилактика

Профилактические мероприятия, позволяющие избежать развития тяжелых осложнений у больных с синдромом Вильямса:
- поддержание в крови кальция и холестерина на оптимальном уровне,
- правильное и сбалансированное питание, предотвращающее появление запоров,
- оптимальная физическая нагрузка,
- уменьшение тревожности ребенка,
- полноценный отдых и сон,
- занятия с детьми в спокойной обстановке,
- диспансерное наблюдение у кардиолога, невролога, психиатра, травматолога-ортопеда.
Для того, чтобы адаптация больных в социуме прошла быстро и безболезненно, необходимо их специальное обучение, внимание и поддержка окружающих лиц, своевременная диагностика и адекватное лечение болезни. Эти мероприятия позволяют повысить уровень жизни пациентов и предупредить неприятные последствия
Общение больных детей со здоровыми сверстниками также оказывает благоприятное воздействие на дальнейшее развитие патологии.
Умственная отсталость препятствует нормальному функционированию организма и жизнедеятельности больных. На основании имеющейся задержки в умственном развитии психиатры присваивают больным инвалидность.
Лечение
Синдром Ангельмана – неизлечимая болезнь, но сегодня существует множество вариантов поддерживающей терапии. Благодаря ей интенсивность некоторых симптомов значительно уменьшается. Справиться с патологией помогают:
- противосудорожные препараты, которые уменьшают количество эпилептических приступов и позволяют управлять ими;
- мелатонин, улучшающий и продлевающий сон;
- лекарства для профилактики гастроэзофагеального рефлюкса у детей раннего возраста;
- поведенческая и коммуникативная терапия, повышающие качество жизни больных;
- трудотерапия для обучения самообслуживания (для больных детей предпочтительна одежда без застежек, пуговиц, обувь без шнурков);
- физиотерапия;
- хирургическое вмешательство при косоглазии;
- ношение брекетов.
Синдром Ангельмана не является прогрессирующим заболеванием. При должной терапии симптомы уменьшаются, но в любом случае больные нуждаются в пожизненном уходе.
Если вы заметили у своего ребенка странности в развитии и поведении, обсудите это со своим педиатром
При осмотре ребенка специалист должен проявить осторожность, так как высока вероятность постановки ошибочного диагноза
Среди особенностей врач должен обратить внимание в первую очередь:
- на характерные аномалии головы и лица;
- счастливую улыбку;
- пропущенные или отсроченные этапы развития ребенка, особенно отсутствие речи;
- моторную дисфункцию (мелкую дрожь, взмах рук, жесткую походку);
- историю развития с судорогами.

Безусловно, ранняя диагностика позволит использовать все терапевтические возможности, которые помогут улучшить жизнь вашего ребенка!
Похожие посты
Оставить комментарий
Симптомы и признаки
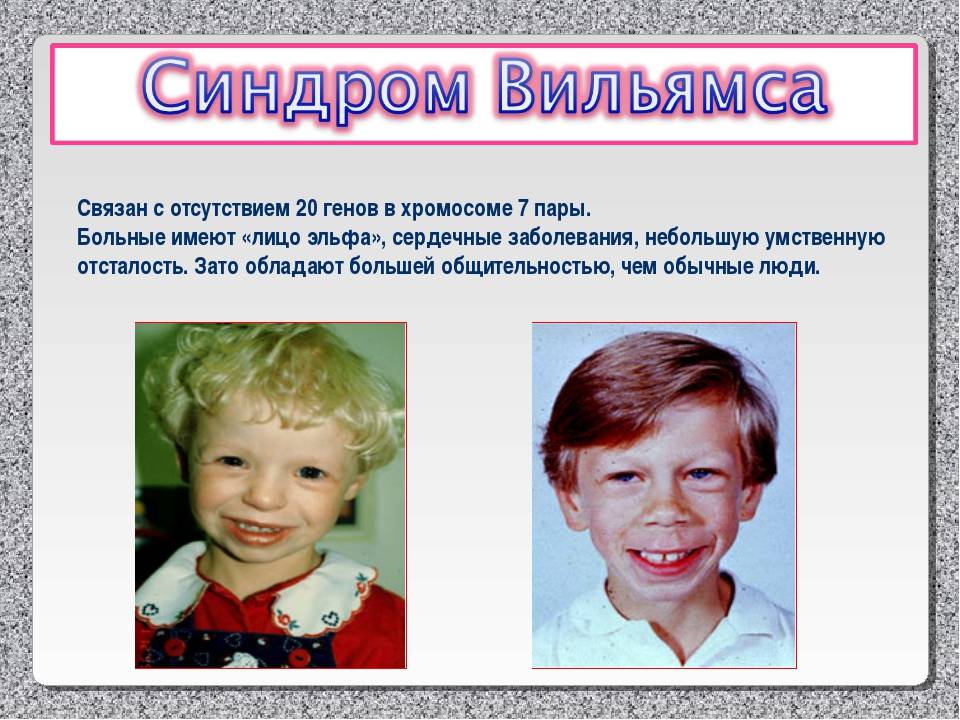
Ребенка с синдромом Вильямса можно распознать по характерной эльфийской внешности, низкому голосу, синеватому оттенку склер.
У многих детей присутствует плоскостопие, что влияет на походку: при ходьбе ребенок немного наклоняется вперед.

У большинства детей обнаруживаются следующие отклонения:
- Патологии сердечно-сосудистой системы. У них часто обнаруживается надклапанный аортальный стеноз, стенозы крупных артерий. Степень выраженности нарушения может варьироваться в зависимости от особенностей заболевания. Также присутствует клапанная недостаточность сердца и другие отклонения.
- Избыточная концентрация кальция. Наблюдается у большинства детей с синдромом Вильямса, часто исчезает самостоятельно в первые годы жизни, но может сохраняться десятилетиями (если это произошло, это нарушение постепенно усугубляет течение заболеваний внутренних органов, характерных для синдрома).
- Умственная отсталость. Выражена средне, уровень IQ находится в границах 40-80, в редких случаях поднимается практически до нормальных показателей. Максимально зафиксированный уровень IQ у человека с синдромом Уильямса — 110. Частично компенсируется особенностями поведения ребенка.
Первые слова ребенок говорит в 2-3 года, а способность соединять их в осмысленные фразы развивается еще позже — после 4 лет.
Нарушения в процессе набора веса. Младенцы с этой патологией медленно набирают вес, в дальнейшем отмечается умеренное отставание в физическом развитии. Их рост редко достигает 160 сантиметров.
Трудности с кормлением. Младенцы часто срыгивают, плохо берут грудь из-за нарушений в мышечной системе.
Болезни желудочно-кишечного тракта. Могут возникать выпячивания в кишечных стенках, которые становятся причиной хронических запоров, также часто наблюдается хронический гастрит типа С.
 Нарушения гормонального фона. Если в процессе внутриутробного развития щитовидная железа не сформировалась, это приводит к возникновению гипотиреоза. Также люди с синдромом Вильямса склонны к развитию сахарного диабета.
Нарушения гормонального фона. Если в процессе внутриутробного развития щитовидная железа не сформировалась, это приводит к возникновению гипотиреоза. Также люди с синдромом Вильямса склонны к развитию сахарного диабета.
Недоразвитость почек, которая приводит к возникновению мочекаменной болезни. Наблюдаются выпячивания в стенках мочевого пузыря.
Косоглазие, обычно сходящегося типа.
Повышенная слуховая чувствительность. Дети чувствительны к звукам, болезненно реагируют на те звуки, которые для обычных людей не создают дискомфорта.
Проблемы с мышечной системой и суставами. Мышечная система ослаблена, а суставы нестабильны, что приводит к прогрессированию нарушений в костных структурах. Отмечаются отклонения в моторике и координации.
Нарушения в костной системе. Характерно возникновения плоскостопия, различных искривлений позвоночного столба, деформации костей в зоне грудной клетки, вальгусное искривление нижних конечностей (колени соединяются).
Заболевания зубов. Отмечаются нарушения прикуса, риск развития кариеса и прочих проблем с зубами повышен.
Часть упомянутых нарушений могут отсутствовать у отдельно взятого ребенка с патологией, но ключевые нарушения, включая специфическую внешность и умственную отсталость, всегда присутствуют.
Также присутствуют характерные интеллектуальные и психологические особенности:
Дети с синдромом крайне дружелюбны, стремятся к общению, выглядят милыми и обаятельными, умеют хорошо разговаривать, чем привлекают к себе внимание. Вежливы, обходительны, улыбчивы
В первые годы жизни тревожно относятся к незнакомцам, но позднее эта проблема сглаживается.
Они практически не могут подмечать мимические особенности лиц других людей, говорящие о том, что человек относится к ним негативно. Это (как и чрезмерное дружелюбие) связано с отклонениями в развитии миндалевидного тела.
Им интересна музыка, они хорошо ее чувствуют и способны добиться успеха в этой сфере.
Несмотря на наличие интеллектуальных дефектов, подросшие дети имеют хороший словарный запас, но многие слова ими могут использоваться неправильно.
Склонны к перепадам настроения, тревожны, может наблюдаться недержание мочи.








Прогноз и профилактика
Продолжительность жизни пациентов с синдромом Вильямса несколько ниже, чем у других людей. Проблемы со здоровьем заключаются в основном в патологии сердца и сосудов и нарушении обмена веществ.
Родители «особых» детей должны знать о случаях внезапной смерти детей с синдромом Вильямса, которые чаще возникают в возрасте до 6 лет. Причиной летального исхода служит острая ишемия миокарда и желудочковая аритмия
Поэтому регулярное обследование сердечно-сосудистой системы и своевременное лечение патологий очень важно для сохранения здоровья малыша.
Дети с генетическим синдромом отстают в развитии от своих сверстников, но их живой характер и покладистый нрав дают возможность малышу социализироваться
Общение с другими детьми очень важно для развития необычного ребёнка и родителям стоит приложить максимум усилий, чтобы помочь малышу адаптироваться в обществе. Взрослые пациенты не могут заниматься трудовой деятельностью из-за своего заболевания и требуют присмотра и контроля со стороны родственников на протяжении всей жизни
Профилактических мер по предупреждению синдрома не существует, поскольку в большинстве случаев причиной патологии является спонтанная мутация. Если же больной генетическим синдромом человек решает завести ребёнка, риск рождения малыша с патологией достигает 50%. В таком случае требуется медико-генетическое консультирования семьи.
Тестирование крови
Этот тест выполняется путем взятия 5 мл крови у ребенка с подозрением на синдром Уильямса. «Флуоресцентное тестирование in situ гибридизации (FISH)» — это тип специализированного хромосомного анализа с использованием специально подготовленных зондов эластина. Если у пациента есть две копии гена эластина (по одной на каждой из их хромосом № 7), у них, вероятно, нет WS. Если у человека есть только одна копия, диагноз WS будет подтвержден «. в соответствии с Ассоциацией синдрома Уильямса. Результаты этого теста могут занять несколько недель, и он проводится только в специализированных лабораториях, поэтому ваш лечащий врач должен убедиться, что тест может быть выполнен до того, как кровь будет взята и отправлена.
Причины
Факторы развития синдрома Ангельмана продолжают исследоваться. Генетический дефект обнаружен в 15 хромосоме материнского набора, но его характер и способ возникновения могут различаться. Иногда заболевание дебютирует в результате передачи измененной генетической информации от родителя, иногда является последствием спонтанных нарушений в геноме. Хромосомные аномалии удается определить примерно у 85-88% больных. Причиной синдрома может быть:
- Делеция. При данном дефекте часть генетического материала теряется или инактивируется. У 70% пациентов диагностируются обширные делеции области 15q12 хромосомы, в которой локализован активатор гена.
- Однородительская дисомия. ОРД определяется в 2-3% случаев болезни. В хромосомном наборе присутствуют две копии 15 хромосомы отца. Материнской хромосомы нет, ген также отсутствует.
- Дефект запечатления. Суть аномалии заключается в том, что центр запечатления, регулирующий активность локуса UBЕ3A, оказывается нефункциональным, «выключенным». Ген остается структурно целым, но не выполняет своих функций. Распространенность ДЗ – 3-5%.
- Мутация UBE3A. У 5-10% пациентов причиной болезни являются мутационные изменения гена. Они представлены инверсиями, микроделециями, транслокациями и дупликациями.
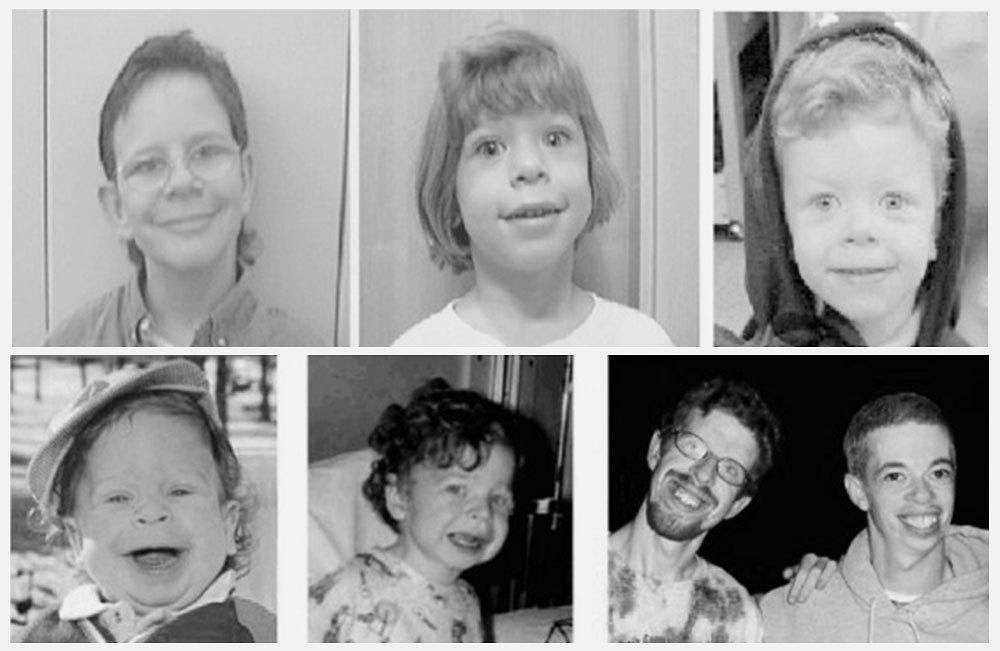
Симптомы синдрома Вильямса
Проявления синдрома Вильямса иногда могут не определяться при рождении ребенка – в этот период заподозрить наличие заболевания можно лишь по сниженной массе тела, врожденному подвывиху бедра (наблюдается не у всех больных), порокам сердца. Все эти факторы достаточно неспецифичны, поэтому чаще всего патологию обнаруживают в старшем возрасте. Одним из самых заметных симптомов синдрома Вильямса является характерный внешний вид лица больных («лицо эльфа») – плоская переносица с округлым носом, увеличенный рот с приподнятыми вверх уголками, пухлые губы, полные щеки, небольшой заостренный подбородок. Из других изменений в области лица и головы часто отмечают низко посаженные уши, выступающий затылок, эпикантус. Для больных синдромом Вильямса характерен голубой цвет глаз с выраженным рисунком на радужной оболочке, голубоватый оттенок склер, отечность верхних и нижних век, нередко – косоглазие.
 Геморрой в 79% случаев убивает пациента
Геморрой в 79% случаев убивает пациента
Из патологических изменений при синдроме Вильямса чаще всего регистрируются различные пороки сердца, что в некоторых случаях может привести к раннему летальному исходу. В основном эти нарушения сводятся к недостаточности различных клапанов. Также у больных синдромом Вильямса чаще, чем у здоровых детей, возникают пупочные и паховые грыжи. Для этого заболевания характерна генерализованная мышечная гипотония, обуславливающая вторичные нарушения формирования скелета – Х-образные ноги, сколиоз и другие искривления позвоночного столба, плоскостопие, деформации грудной клетки. При осмотре стоматолога у больных синдромом Вильямса выявляются длинные, но редко расположенные зубы, которые довольно часто поражаются кариесом и легко разрушаются.
Дети, страдающие синдромом Вильямса, значительно отстают от сверстников, как в интеллектуальном, так и в физическом развитии. Они имеют пониженную массу тела при рождении, медленно набирают ее в дальнейшем, их рост останавливается намного раньше, поэтому проявлением этого заболевания также является низкорослость. Масса тела после подросткового периода может увеличиваться, имеется риск развития ожирения. В интеллектуальном плане больные синдромом Вильямса на всю жизнь сохраняют признаки имбецильности (умеренная умственная отсталость, средний уровень IQ 40-80). При этом они достаточно легко идут на контакт, у них, как правило, хорошо развита устная речь, при правильной коррекционной работе они способны выполнять простейшие бытовые поручения. В некоторых случаях при синдроме Вильямса может наблюдаться эмоциональная неустойчивость, тревожность, энурез.
Эпидемиология
Около 25% всех смертельных
исходов не связано с неизлечимыми
заболеваниями либо старческими или деструктивными изменениями в мозге. В Европе
ежегодно регистрируется около 700000, а в США – 400000 случаев
внезапной смерти.
В результате реанимации на
догоспитальном и госпитальном этапах примерно в более чем половине случаев
удается восстановить спонтанное кровообращение. Однако 50% из этих пациентов в
последующем умирает, главным образом в результате кардиального или
церебрального повреждения.
Уровень выживаемости пациентов
(число выживших после реанимации пациентов, которые выписались из лечебного
учреждения), перенесших остановку кровообращения в больничных условиях,
колеблется от 0 до 29% (в среднем 14%), а внебольничных — от 0 до 40%.
Основной фактор, влияющий на уровень выживаемости – длительность интервала
времени с момента остановки кровообращения до начала СЛР. Важным
прогностическим фактором исхода СЛЦР является первичный механизм остановки
кровообращения. В возрасте менее 10 лет более высокий уровень выживаемости, чем
старше 10 лет; уровень выживаемости не отличается у пациентов в возрасте 10–70
лет и прогрессивно снижается у лиц старше 70 лет.
Знакомство с болезнью
Синдром Аспергера представляет собой психическое расстройство, характеризующееся, прежде всего, проблемами социальной адаптации и стереотипным поведением, но не сопровождающееся умственной отсталостью и речевым недоразвитием. В отдельных случаях такие больные, наоборот, обладают высоким интеллектуальным уровнем.
Впервые о синдроме услышали в середине 20 века, когда австрийский психиатр, педиатр Ганс Аспергер исследовал и описал четырех детей с проблемами социального взаимодействия. Они испытывали сложности в налаживании бессловесного контакта с людьми, отличались эмоциональной скупостью и неспособностью сопереживать другим, а также проявляли некоторую неуклюжесть в движениях.
Кроме того, симптомы данного расстройства проявлялись и у самого его первооткрывателя.
Обнаруженный синдром врач обозначил как аутистическая психопатия. Он рьяно и подобострастно защищал свои «находки», говоря о том, что люди с аутистической психопатией, пройдя через большие трудности в детстве, занимают определенную ячейку в обществе и вносят свой вклад в его развитие. Ученый считал, что такие больные обладают особым складом мышления и в дальнейшем могут достичь значительных высот.
В 90-х годах расстройству было присвоено его современное название: синдром Аспергера. Понятие выделили в самостоятельный диагноз и сформулировали его основные критерии.
В настоящее время Международная классификация болезней отвергает этот термин. Расстройство зашифровано под кодом «Шизоидное расстройство детского возраста».
Одна из теорий возникновения заболевания основана на наследственном факторе. Считается, что, если у ребенка имеются больные родственники, особенно отец, у него есть повышенный риск заработать расстройство.
Предполагается, что еще внутриутробно под воздействием тератогенных, то есть разрушающих, факторов, у плода формируются аномалии и пороки развития. Они приводят к патологической миграции эмбриональных клеток, вызывающих изменения в структуре головного мозга. Последние, в свою очередь, нарушают нейронные пути, отвечающие за мышление и поведение.








Диагностика
Диагностика синдрома Вильямса не вызывает у специалистов особых затруднений. Она основывается на данных хромосомного анализа, в основе которого лежат генетические тесты, подтверждающие или опровергающие предполагаемый диагноз синдром Вильямса.
Молекулярно-цитогенетическое исследование проводят методом флюоресцентной гибридизации или с помощью ДНК-микрочипа. Флуоресцентная гибридизация предназначена для определения специфической последовательности генов в организме человека. Проводят его перед внедрением оплодотворенной яйцеклетки в стенку матки, во время беременности и сразу после рождения ребенка. ДНК-микрочип – диагностическая методика, которая проводится с помощью ПЦР. На специальный микрочип наносят геном, а затем его исследуют.
Дополнительные исследования:
- общеклинический анализ крови и мочи,
- биохимия крови — гиперхолестеринемия, гиперкальциемия,
- определение гормонального статуса,
- исследование мочи на содержание креатинина и кальция,
- ЭКГ и ЭхоКГ,
- тонометрия,
- УЗИ сердца и почек,
- рентгеноурологическое исследование,
- томография мозга.
Диагностика заболевания
Постановка диагноза основывается на выявлении характерных внешних проявлений больного, изменений зубов и наличия множественного кариеса, нарушения опорно-двигательного аппарата и мышечной гипотонии, отставания в физическом и нервно-психическом развитии. При осмотре нередко можно наблюдать пупочные и пазовые грыжи, сходящееся косоглазие, повышение артериального давления, при разговоре отмечается хриплый голос. Но такие симптомы не являются специфичными, за исключением фенотипических признаков, поэтому требуется дополнительное обследование ребёнка.
На основании клинических проявлений синдром Вильямса не ставится!
Лабораторное исследование
- общий анализ крови позволяет выявить снижение количества нейтрофилов;
- биохимический анализ определяет повышение концентрации кальция и тиреотиопного гормона;
- общий анализ мочи может изменяться при вовлечении в патологический процесс мочевыделительной системы.
Молекулярно-генетический анализ
Для определения локализации дефекта в хромосоме используется молекулярно-генетический анализ, который проводится врачом-генетиком, так называемый – FISH-метод. Такое диагностическое исследование при синдроме Вильямса регистрирует мутацию в хромосоме 7q11.23. Данный анализ может использоваться как метод пренатальной диагностики. Особенно он актуален для оценки риска рождения больного ребёнка в процессе экстракорпорального оплодотворения.

Инструментальные методы диагностики
- эхокардиологическое исследование (ЭХО-КГ) позволяет визуализировать врождённые потоки сердца (недостаточность аортального и митрального клапанов, надклапанные стенозы аорты и лёгочного ствола). У детей более старшего возраста отмечается изменения стенки аорты, сужение коронарных сосудов и кальциноз их стенок и клапанов сердца;
- магнитно-резонансная томография головного мозга (МРТ) наводит на мысль о заболевании, при выявлении недоразвития поражённого органа и изменения размеров некоторых его структур;
- ультразвуковое исследование почек и мочевого пузыря позволяет определить уровень поражения при прогрессировании заболевания;
- аудиограмма для определения остроты слуха.
Консультация врачей-специалистов: кардиолога, ортопеда, невролога, генетика, оториноларинголога и офтальмолога.
Принципы выбора противопаркинсонического препарата
Выбор препарата на начальном этапе лечения проводят с учетом возраста, выраженности двигательного дефекта, трудового статуса, состояния нейропсихологических функций, наличия сопутствующих соматических заболеваний, индивидуальной чувствительности пациента. Помимо достижения оптимального симптоматического контроля, выбор препарата определяется необходимостью отсрочить момент развития моторных флуктуаций и дискинезий (таблица 2).
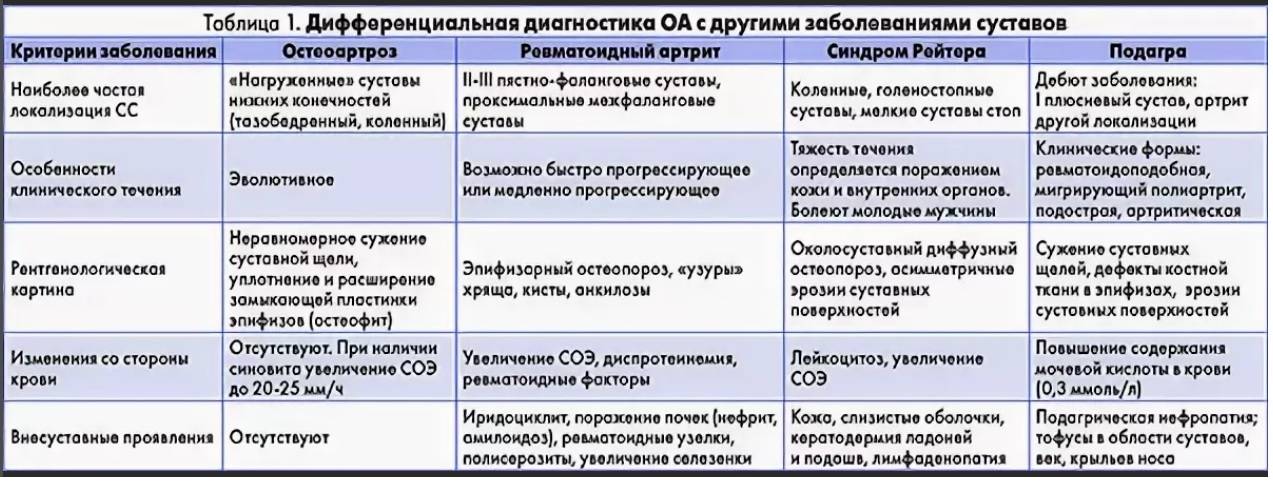
Таблица 2. Выбор препарата для начального лечения болезни Паркинсона.
| Препараты | Возможность использования в качестве средства первого выбора | Степень симптоматического улучшения | Нейро-протективный потенциал | Риск побочного действия | |
| Флуктуации и дискинезии | Другие побочные эффекты | ||||
| Леводопа | + | +++ | +? | ↑ | ↑ |
| Агонисты дофаминовых рецепторов | + | ++ | +? | ↓ | ↑ |
| Ингибитор МАО В | + | + | +? | ↓ | ↑ |
| Амантадин | + | + | +? | ↓ | ↑ |
| Холинолитики | — | + | — | ? | ↑ |
У лиц моложе 50 лет при легкой или умеренной выраженности двигательных нарушений в отсутствие выраженных когнитивных нарушений назначают один из следующих препаратов: агонист дофаминовых рецепторов, ингибитор моноаминооксидазы типа В, амантадин. При более легком двигательном дефекте может быть назначен ингибитор МАО В, при более выраженном дефекте предпочтительнее начинать с лечения с одного из агонистов дофаминовых рецепторов. Неэрголиновые агонисты (например, прамипексол, ропинирол, ротиготин или проноран) ввиду более благоприятного профиля побочных эффектов предпочтительнее, чем эрголиновые (бромокриптин, каберголин). При недостаточной эффективности или плохой переносимости одного из агонистов дофаминовых рецепторов может быть испробован другой агонист дофаминовых рецепторов или препарат другой фармакологической группы. Рациональна комбинация агониста дофаминовых рецепторов, ингибитора МАО типа В и амантадина, к которой следует переходить постепенно, добавляя препарат новой группы, если ранее назначенное средство не обеспечило ожидаемого эффекта.
Антихолинергические средства (например, бипериден) показаны при наличии выраженного тремора покоя либо болезненной дистонии при условии сохранности нейропсихологических функций. Их целесообразно добавлять к комбинации агониста дофаминовых рецепторов с ингибитором МАО В и/или амантадином, если она у пациента относительно молодого возраста не обеспечила подавления тремора в той степени, в которой это необходимо для поддержания его трудоспособности.
Если указанные препараты в максимально переносимых дозах и их комбинация не обеспечивают адекватного состояния двигательных функций и социальной адаптации больных, назначают препарат леводопы в минимальной эффективной дозе .
У лиц в возрасте 50–70 лет при умеренном двигательном дефекте и относительной сохранности когнитивных функций лечение начинают с ингибитора МАО типа В (при легких симптомах паркинсонизма) или одного из агонистов дофаминовых рецепторов. В дальнейшем целесообразен постепенный переход к комбинации агониста дофаминовых рецепторов, ингибитора МАО типа В и амантадина (при условии хорошей переносимости). Больным после 60 лет антихолинергические средства, как правило, не следует назначать из-за риска ухудшения познавательных функций и других побочных эффектов. При недостаточной эффективности комбинации указанных выше препаратов добавляют препарат леводопы в минимальной эффективной дозе (200–400 мг в сутки).
У лиц в возрасте 50–70 лет при выраженном двигательном дефекте, ограничивающем трудоспособность и(или) возможность самообслуживания, а также при наличии выраженных когнитивных нарушений и необходимости получения быстрого эффекта лечение начинают с препаратов, содержащих леводопу. Если небольшие или средние дозы леводопы (300–500 мг леводопы в сутки) не обеспечивают необходимого улучшения, к ним последовательно могут быть добавлены агонист дофаминовых рецепторов, амантадин и ингибитор МАО В.
У пожилых лиц (старше 70 лет), особенно при наличии выраженного когнитивного снижения и соматической отягощенности, лечение следует начинать с препаратов леводопы. Указанные возрастные границы относительны, и общий принцип скорее заключается в том, что чем моложе больной, тем позже следует вводить препараты леводопы. Кроме того, решающее значение играет не столько хронологический, сколько биологический возраст больных.
Лечение
- Для нормализации артериального давления больным назначают гипотензивные препараты из группы ингибиторов АПФ, антагонистов кальция, диуретиков, бета-адреноблокаторов.
- Ноотропные препараты для улучшения мозгового кровообращения и адекватного психического развития больных – «Пирацетам», «Винпоцетин», «Церебролизин».
- При высоком уровне кальция в крови требуется его медикаментозная коррекция — внутривенное введение физраствора, диуретики, препараты калия.
- При развитии тяжелых форм патологии назначают глюкокортикостероиды – «Гидрокортизон», «Преднизолон».
- Физиотерапевтические процедуры улучшают подвижность суставов — ЛФК, магнитотерапия, ультразвук, электротерапия, акупунктура, массаж.
- Психологическая коррекция заключается в обучении больных детей правильной речи, чтению и письму, уменьшении тревожности и страха.
- Дружелюбие и взаимопонимание в семье играют важную роль в лечении больных детей.
- Психотерапия и занятия с психологом — неотъемлемая часть лечебного процесса.
- Хирургическое вмешательство направлено на устранение у детей врожденных пороков сердца, нередко приводящих к смерти больных.
Общая информация о патологии
У большинства больных нарушения в работе сердца и сосудов, которые представлены аорты и артерий различной степени тяжести. При уменьшении просвета почечной артерии половина пациентов страдает высокими показателями артериального давления.
В кровеносном русле наблюдаются значительные количественные показатели кальция, что вызывает психоэмоциональные изменения и болезненное вздутие живота на протяжении первых месяцев жизни.
Пациенты, страдающие заболеванием, имеют низкую массу тела. Набирают они ее намного медленнее, чем их здоровые сверстники. Первый год жизни очень тяжелый по поводу вскармливания. У детей частые позывы на рвоту, нарушены процессы сосания и глотания.
Также могут наблюдаться:
- патологии желудка и кишечного тракта;
- гипофункция щитовидной железы;
- дефекты строения почек;
- грыжи различной локализации;
- низкий тонус мышц.