Обзор синдрома Марфана

Синдром Марфана — это генетическое заболевание, при котором изменяются белки, которые помогают формировать здоровую соединительную ткань. Это приводит к проблемам с развитием соединительной ткани, которая поддерживает кости, мышцы, органы и ткани вашего тела. Мутации (изменения) определенного гена вызывают синдром Марфана, большинство людей наследует это заболевание от своих родителей.
Симптомы синдрома Марфана могут проявляться как в легких, так и тяжелых формах и различаться, поскольку данное заболевание может влиять на разные области тела, в том числе:
- Скелет, который включает кости и соединительные ткани, такие как связки, сухожилия и хрящи.
- Глаза.
- Сердце и кровеносные сосуды, включая вены, артерии и клапаны внутри сердца.
- Кожа.
- Легкие.
Лечение варьируется в зависимости от пораженной области тела и может включать в себя лекарства, другие методы лечения и хирургическое вмешательство для лечения состояния и его осложнений. Исследования и достижения в области лечения и операций позволяют людям с синдромом Марфана жить долгой и продуктивной жизнью.
Синдром Марфана у детей и взрослых, сопровождающийся аномалией развития зрительного аппарата
Различные поражения органов зрения при данной болезни диагностируются у 50-80% пациентов, причем нарушения начинают проявляться уже в детском возрасте. Это могут быть следующие патологии:
- эктопия хрусталика;
- сильная близорукость;
- уплощение и увеличение размера роговицы;
- косоглазие;
- гипоплазия радужной оболочки и цилиарной мышцы;
- изменения в сетчатке;
- деструкция стекловидного тела и другие.
Терапия глазных заболеваний проводится обычными в таких случаях методами. Однако их воздействие может быть затруднено текущими процессами в организме. Рассмотрим подробнее офтальмологические патологии, сопутствующие синдрому Марфана.
Обследование и постановка диагноза
Врач запишет историю болезни и проведет физикальный осмотр. Осмотр может включать:
Тест с наклоном вперед. Врач попросит вас наклониться вперед, наблюдая за вашим позвоночником со стороны. При кифозе искривление верхней части спины может стать более заметным в этом положении. При постуральном кифозе дефекты корректируются сами, когда вы лежите на спине.
Тест на проверку неврологических функций
Хотя неврологические изменения, сопровождающие кифоз, являются редким явлением, врач может проверить их наличие, обращая внимание на слабость, изменения в чувствительности или паралич ниже места кифоза.. Визуализация позвоночника
Врач может назначить рентгенологическое обследования для подтверждения наличия кифоза, определения степени искривления и выявления деформации позвонков, что помогает определить вид кифоза. Например, внешний вид клинообразных позвонков или другие характеристики, видимые на рентгеновских снимках, помогают провести различие между постуральным кифозом и кифозом Шейермана. У взрослых рентген может выявить артритные изменения в позвоночнике, которые могут привести к усилению боли. Если врач подозревает наличие опухоли или инфекции, он может рекомендовать проведение МРТ позвоночника.
Визуализация позвоночника. Врач может назначить рентгенологическое обследования для подтверждения наличия кифоза, определения степени искривления и выявления деформации позвонков, что помогает определить вид кифоза. Например, внешний вид клинообразных позвонков или другие характеристики, видимые на рентгеновских снимках, помогают провести различие между постуральным кифозом и кифозом Шейермана. У взрослых рентген может выявить артритные изменения в позвоночнике, которые могут привести к усилению боли. Если врач подозревает наличие опухоли или инфекции, он может рекомендовать проведение МРТ позвоночника.
Исследование функции легких. Врач может также провести исследование дыхательных функций, чтобы оценить проблемы с дыханием, вызванные кифозом.
Причины
Позвоночник (позвоночный столб) состоит из костей (позвонков), которые соединены плотными, фиброзными связками. Позвоночник состоит из 7 шейных (цервикальных) позвонков, 12 позвонков грудного отдела (торакальных) и 5 поясничных (люмбальных) позвонков. Поясничные позвонки – самые крупные, они удерживают большую часть массы тела. Крестец, состоящий из 5 сросшихся позвонков, расположен ниже поясничных позвонков. Три последних маленьких позвонка, также сросшиеся вместе, формируют копчик.
Причины кифоза различаются по типам этого заболевания.
Разновидности кифоза у детей и подростков
Постуральный кифоз (осаночный кифоз). Эта форма кифоза главным образом встречается у подростков. Развитие постурального кифоза обычно происходит медленно. Он более типичен для девочек, чем для мальчиков. Плохая осанка или сутулость могут вызвать растяжение позвоночных связок и формирование несоответствующих норме костей позвоночника (позвонков). Постуральный кифоз часто сопровождается чрезмерно вогнутым изгибом (гиперлордоз) в нижнем (поясничном) отделе позвоночника. Гиперлордоз – это способ компенсирования организмом чрезмерно выгнутого изгиба в верхней части позвоночника.
Кифоз Шейерманна. Как и постуральный кифоз, кифоз Шейермана обычно развивается у подростков в возрасте 10-15 лет, пока кости находятся в процессе роста. Данный кифоз также называется болезнью Шейермана-Мау, он встречается в два раза чаще у мальчиков, чем у девочек. При болезни Шейермана-Мау позвонки деформируются таким образом, что на рентгеновских снимках они выглядят клиновидными, а не треугольными. На пораженных позвонках также могут развиться грыжи Шморля. Эти грыжи появляются в результате продавливания подушки (диска), расположенной между позвонками, в кость в нижней и верхней части позвонка.
Причина возникновения кифоза Шейермана не известна, но обычно он является наследственным. У некоторых пациентов, страдающих данной формой кифоза, также развивается сколиоз – деформация позвоночника, которая вызывает боковой изгиб. Взрослые, у которых болезнь Шейермана-Мау развивалась в детстве, могут с возрастом испытывать усиливающуюся боль.
Врожденный кифоз. Врожденный порок позвоночника, формирующийся во время развития плода, вызывает кифоз у некоторых младенцев. Могут срастись несколько позвонков или могут неправильно сформироваться кости. Эта форма кифоза может усугубиться в процессе роста ребенка. Кифоз является самой распространенной причиной паралича нижней части тела (параплегии) после травмы и инфекции.
Причины возникновения у взрослых
Заболевания, которые могут вызвать искривление позвоночника у взрослых, что приводит к кифозу, включают:
- Остеопороз – заболевание, связанное со снижением плотности костей, часто приводящее к переломам позвонков, что вызывает сдавливание позвоночника и развитие кифоза;
- Дегенеративный артрит позвоночника, который может вызвать разрушение костей и дисков позвоночника;
- Анкилозирующий спондилоартрит (болезнь Бехтерева), воспалительный артрит, поражающий позвоночник и близлежащие суставы;
- Нарушения со стороны соединительной ткани, такие как синдром Марфана, которые влияют на способность соединительной ткани удерживать суставы в правильном положении;
- Туберкулез и другие инфекции позвоночника, которые могут привести к разрушению суставов;
- Рак или доброкачественные опухоли, которые воздействуют на кости позвоночника и обуславливают изменение их положения;
- Spina Bifida – это врожденный дефект. Тот отдел позвоночника, в котором он расположен, формируется не полностью, он вызывает поражение спинного мозга и позвонков;
- Состояния, вызывающие паралич, например, корковый паралич и полиомиелит, а также те состояния, в результате которых снижается гибкость костей позвоночника.
3.Симптомы и диагностика
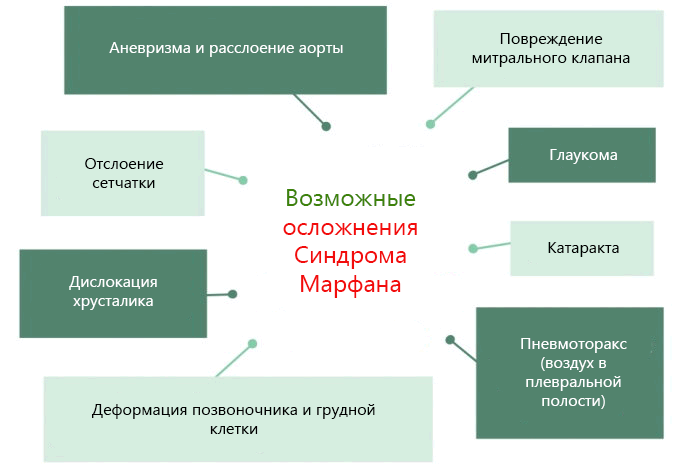
Синдром Марфана обнаруживается уже в раннем возрасте; он может прогрессировать или оставаться относительно стабильным. Классической триадой считают деформацию опорно-двигательного аппарата, офтальмологическую патологию и сердечнососудистую недостаточность.
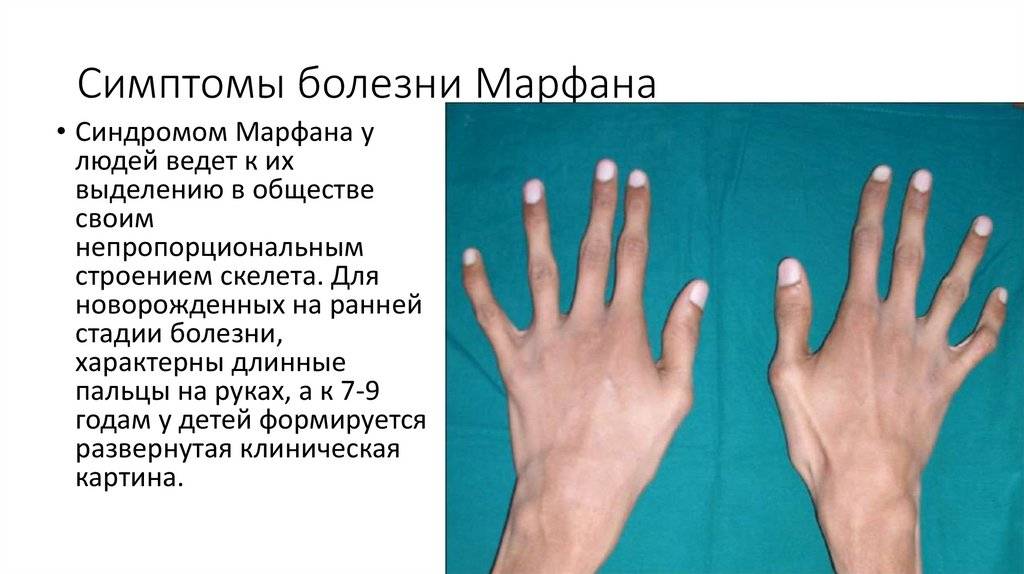
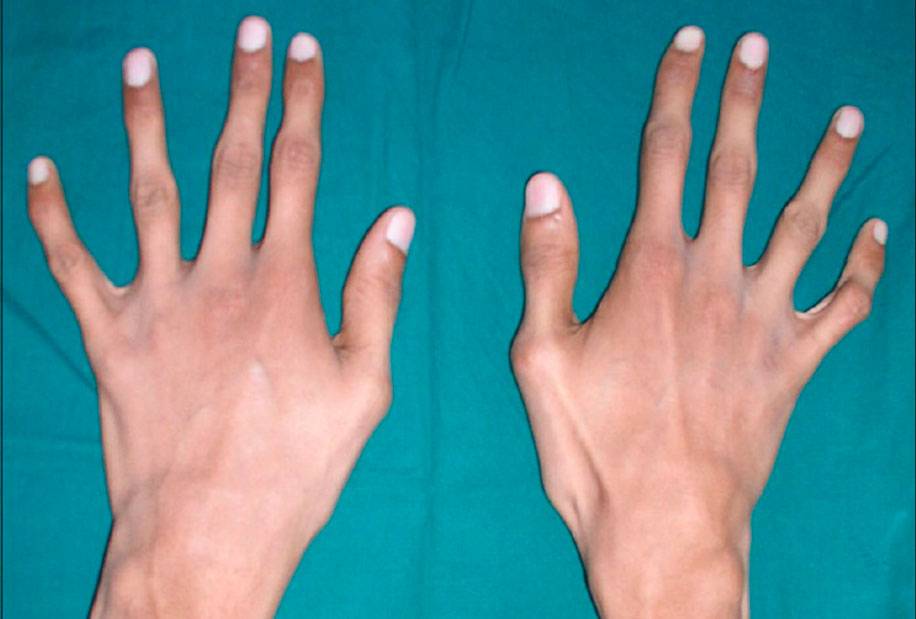
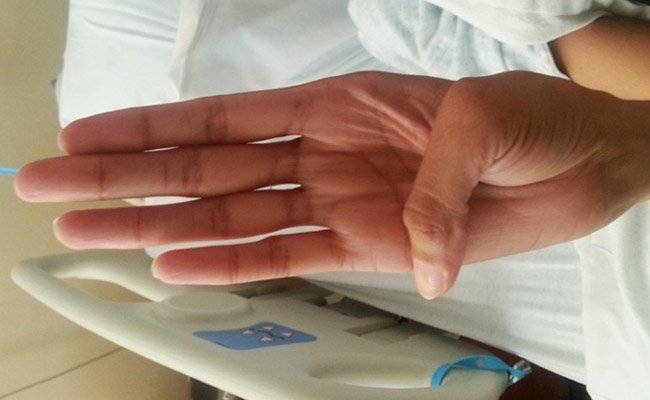
Больные отличаются высоким ростом и диспластичным телосложением: непропорционально длинные конечности, «паучьи пальцы» (арахнодактилия), килевидная или вдавленная грудная клетка, сложные искривления позвоночника (лордоз, кифосколиоз), вальгусная или варусная конфигурация ног, патологическая подвижность суставов, плоскостопие и т.д.
Характерные диспропорции обнаруживаются также в строении челюстно-лицевых структур. Явно недостаточно развиты мышечная и жировая ткани. Типичным признаком является аневризма аорты и/или других крупных сосудов, в т.ч. легочных и мозговых; встречается множество различных пороков сердца. Столь же полиморфными и тяжелыми могут быть аномалии развития органов дыхания, пищеварения, а также центральной нервной и зрительной систем (в частности, офтальмологически выявляется выраженная миопия или гиперметропия, подвывих хрусталика, полное отсутствие радужки или хрусталика, удлинение глазного яблока и т.п.).
Комплекс столь критичных по сути аномалий может быть выражен относительно мягко, однако в целом синдром Марфана обычно нуждается в квалифицированном терапевтическом наблюдении и сопровождении, – в противном случае и качество, и продолжительность жизни резко ограничиваются (в отсутствие курации непосредственной причиной смерти становится, как правило, хроническая или острая, наступающая вследствие разрыва аневризмы, сердечная недостаточность).
Диагноз устанавливается путем изучения семейного анамнеза и всестороннего обследования: практически любой из существующих сегодня диагностических методов выявляет те или иные характерные изменения.
Основные положения
Причиной синдрома Марфана является аутосомно-доминантная мутация гена, кодирующего гликопротеин фибриллин-1, который является основным компонентом микрофибрилл, вследствие чего возможны многочисленные деформации и дефекты.
Проявления варьируют в широких пределах, но основные структурные дефекты затрагивают сердечно-сосудистую и опорно-двигательную системы, а также орган зрения, в результате чего типичным является сочетание удлиненных конечностей, расширения корня аорты и дислокации хрусталиков.
Расслоение аорты является наиболее опасным осложнением.
Диагностика основывается на клинических критериях; часто проводится генетическое исследование.
Для выявления структурных аномалий необходимо провести диагностическую визуализацию органов скелетной и сердечно-сосудистой систем и органа зрения.
Всем пациентам следует назначить бета-блокаторы для профилактики аортальных осложнений; лечение других осложнений следует проводить по мере их возникновения.
Лечение синдрома Марфана
От синдрома Марфана полностью избавиться и устранить механизм его развития невозможно. Лечение базируется на улучшении общего состояния больного, устранении клинических проявлений и проведении профилактических мероприятий, препятствующих развитию осложнений.








Больным с данным синдромам рекомендовано ограничить физическую нагрузку до низкого уровня, или минимального. Риск появления патологии сердечно-сосудистой системы возрастает при средних и высоких физических нагрузках.
Следует обходить стороной и повседневные нагрузки, при которых возможно повышение внутригрудного давления, ведущему к развитию пневмоторакса (например, подъем тяжестей, подъем по этажам).
Медикаментозное лечение
Медикаментозная терапия направлена на устранение клинической картины заболевания.
Со стороны сердечно-сосудистой системы рекомендованы β-адреноблокаторы (например: Анаприлин), которые снижают скорость распространения пульсовых волн при стремительно растущем расширении аорты и обратного тока крови на двустворчатом клапане или клапане аорты.
β-адреноблокаторы также оказывают положительный эффект при нарушении ритма и проводимости, в сочетании с сердечными гликозидами.
Но следует помнить о существующих противопоказаниях данных групп препаратов:
- хронический обструктивный бронхит;
- бронхиальная астма;
- снижение частоты сердечных сокращений;
- низкое артериальное давление.
Блокаторы каналов кальция используются при наличии противопоказаний к В-адреноблокаторам.
Хирургия
Хирургическое лечение проводится, если есть осложнения со стороны сердечно-сосудистой системы, с целью коррекции пораженных участков. Его проводят при пролабировании двустворчатого клапана и расслаивании аорты.
При этом осуществляется протезирование двустворчатого клапана.
У беременных с тяжелым течением болезни Марфана, роды разрешаются хирургическим путем.
Синдром Марфана у детей
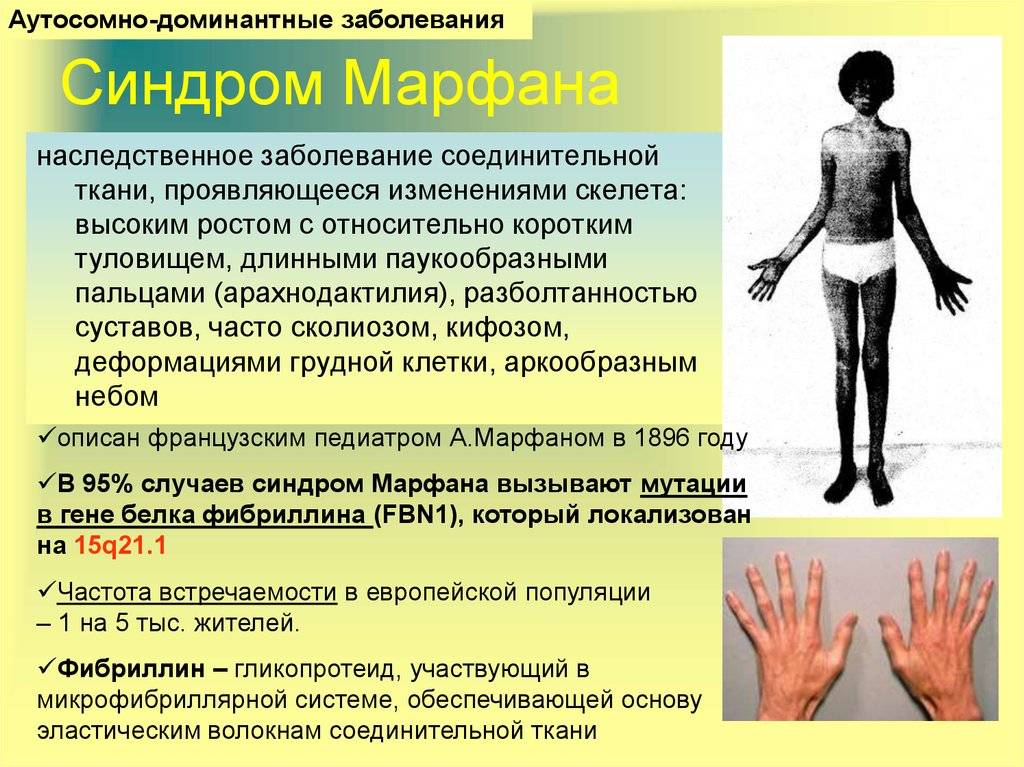
Синдром Марфана (врожденная мезодермальная дистрофия) — это аутосомно-доминантное генетическое заболевание соединительной ткани с преимущественным поражением скелета (удлинение трубчатых костей, долихостеномелия, арахнодактилия, гипермобильность суставов), глаз (миопия, подвывих хрусталика) и сердечно-сосудистой системы (пролапс митрального клапана, расслаивающая аневризма аорты). Впервые данная патология была описана в 1896 году французским педиатром А. Марфаном, который выявил характерную деформацию скелета у 5-летней девочки (длинные трубчатые кости, паукообразные пальцы рук, высокорослость). Болезнь Марфана возникает в результате нарушения синтеза фибриллина-1, который является основным структурным белком соединительной ткани. Данный синдром обусловлен мутацией гена, кодирующего продукцию этого гликопротеина.
Синдром Марфана у детей относится к редким врожденным аномалиям. Частота диагностированных случаев составляет 1:10 000 – 1:20 000. От болезни Марфана чаще страдают мальчики.
Для данной патологии характерны следующие признаки:
- аутосомно-доминантный тип наследования (если один из родителей имеет данное заболевание, то риск возникновения его у ребенка колeблется от 50 до 100%);
- высокая пенетрантность (болезнь фенотипически проявляется при небольшом количестве копий аллелей мутантного гена);
- различная экспрессивность (трaнcформация гена в определенную белковую структуру).
Кроме семейного наследования, синдрома Марфана в 25% случаев возможна первичная мутация гена, что объясняет рождение больного ребенка у здоровых родителей.
Среди известных мировых личностей синдромом Марфана болели Авраам Линкольн, Ганс Христиан Андерсен, Сергeй Рахманинов, а американскому пловцу Майклу Фелпсу данный генетический порок дал большое преимущество перед другими спортсменами, что позволило ему стать 23-кратным олимпийским чемпионом. Некоторые ученые утверждают, что люди с болезнью Марфана владеют уникальным интеллектом. Данный факт стал основанием для поиска корреляционной связи между геном данного заболевания и геном «гениальности».
Патологии сетчатки при синдроме Марфана
Из-за слабости соединительной ткани подвержена растяжению также сетчатка глаза. В результате этого повышается риск появления периферических хориоретинальных дистрофий — локальных истончений сетчатки, которые могут спровоцировать отторжение слоя светочувствительных клеток от пигментного эпителия. Такое нарушение очень опасно: начинает падать острота зрения, ухудшается восприятие света и цвета.
Признаками отслойки сетчатки могут быть следующие проявления:
- вспышки, искры в глазах — такое явление называется фотопсией;
- искажение формы, размера и оттенка объектов — метаморфопсия;
- «мушки» и черные точки перед глазами как следствие повреждения ретинального сосуда;
- выпадение из поля зрения отдельных элементов в видимой картинке — это признак того, что отслоение началось в центральной зоне сетчатки;
- появление темной пелены, охватывающей все большую область, снижение периферийной видимости.
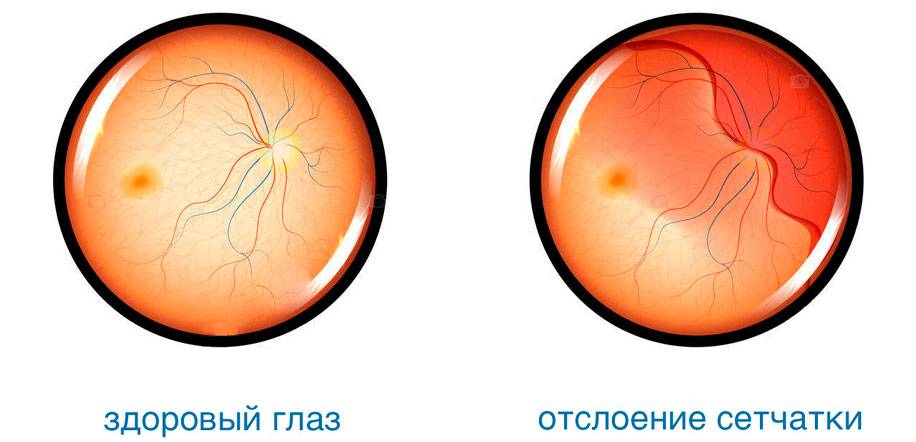
Отслоение сетчатки сегодня поддается успешному лечению различными методами. Наиболее эффективным из них является лазерная коагуляция — прижигание поврежденных участков с целью надежного их соединения с сосудистой оболочкой.
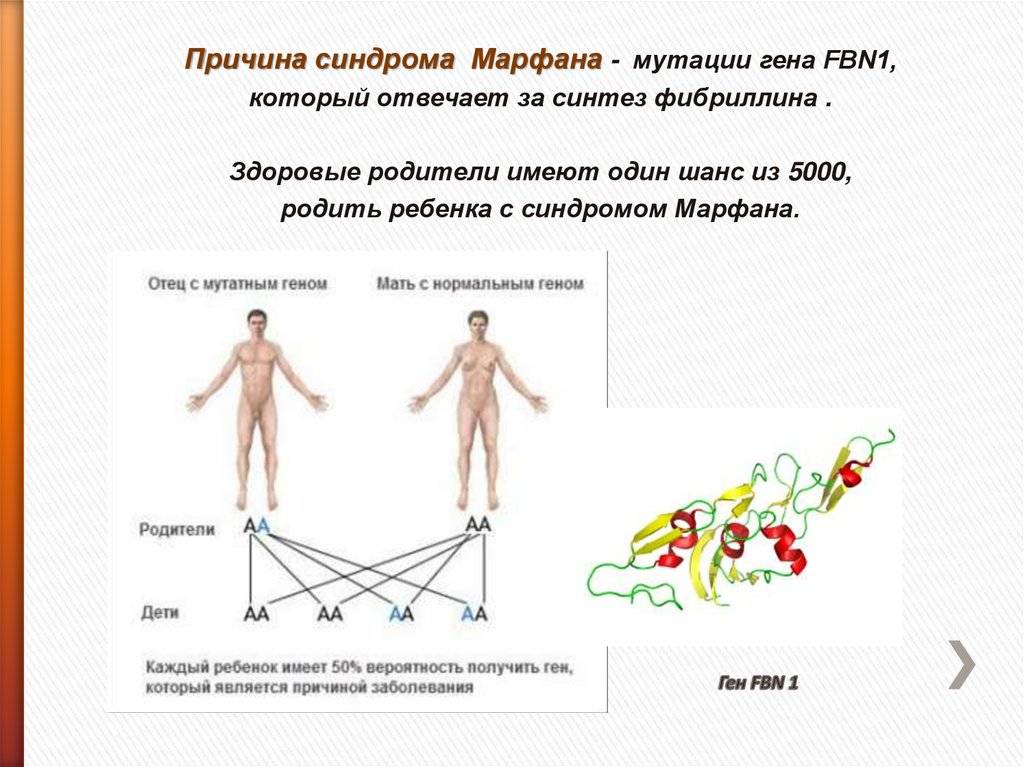
Причины синдрома Марфана
Данное генетическое заболевание вызвано дефектом гена FBN1 в длинном плече 15 хромосомы. Этот ген кодирует белок гликопротеин фибриллин-1, который отвечает за прочность и эластичность соединительной ткани. Соответственно, все проявления патологии связаны с тем, что соединительнотканные структуры в организме человека теряют свои нормальные свойства.
Наследуется мутация по аутосомно-доминантному признаку, то есть дети получают патологический ген от родителей, которые страдают от патологии. При этом шанс ребенка получить мутацию от одного из родителей составляет 50% (рис. 1). Синдром не передается через поколение: здоровые дети больных родителей не могут передать ген своим потомкам.
Однако примерно у 25% людей с синдромом Марфана никто из родителей не оказывается носителем аномалии гена FBN1: в таком случае мутация развивается спонтанно.
До сих пор не выявлено определенных факторов риска развития этого генетического нарушения: заболевание встречается одинаково часто среди мужчин и женщин, а его распространенность не зависит от расы или этнической группы. Частота заболеваемости у этой патологии составляет примерно 1 случай на .
Если клинические признаки мутации ярко выражены, заподозрить болезнь можно уже в первые месяцы жизни ребенка, но стертые формы заболевания часто проявляются уже во взрослом возрасте, когда пациент обращается к врачам по поводу различных проявлений синдрома.
Важно! Не стоит записываться на генетическое обследование в качестве медосмотра. Поиски «поломки» гена FBN1 оправданы только в случае, если болезнь проявляет себя характерными признаками: бессимптомное носительство этой мутации невозможно
Если у одного из родителей установлен этот диагноз, будущей маме следует пройти генетическое обследование еще до родов. Это позволит заранее узнать, передалась ли аномалия ребенку.









Жизнь с синдромом Марфана
Лечение синдрома Марфана и его осложнений и жизнь с ним — это процесс на всю жизнь. Однако успехи в лечении позволяют людям с этим расстройством вести долгую и продуктивную жизнь. Следующие советы помогут вам лучше справляться с этим расстройством:
- Регулярные посещения врача важны при лечении синдрома Марфана и предотвращения осложнений. Посещения могут включать регулярные осмотры глаз, визуальные тесты для проверки на наличие проблем с сердцем и легкими, а также оценку вашего скелета и роста.
- Обратитесь за поддержкой. Поговорите со своей семьей и друзьями о расстройстве и своих чувствах. Подумайте о том, чтобы присоединиться к сообществу или группе поддержки в Интернете.
- Обратитесь за консультацией или поговорите со специалистом в области здравоохранения, если вы чувствуете депрессию или беспокойство по поводу синдрома Марфана и его последствий для вашего тела.
- Придерживайтесь здоровой, сбалансированной диеты, богатой фруктами, овощами и цельнозерновыми продуктами.
- Если вы курите, бросьте. Курение может негативно сказаться на здоровье ваших костей и легких.
Женщины с синдромом Марфана могут иметь здоровую беременность. Тем не менее, беременность сопряжена с высоким риском, поскольку она может увеличить нагрузку на сердце. Если вы думаете о беременности, поговорите со своим врачом. Планирование помогает врачам лечить проблемы до беременности, чтобы сохранить здоровье матери и ребенка.
Просмотров 133, за сегодня 1
Почему проявляется генетический синдром?
Причиной развития недуга считается мутация в гене FBN1, который располагается в 15 хромосоме и отвечает за нормальное производство фибриллина 1. Этот белков соединительной ткани является одним из главных компонентов, придающих ей эластичность и способность к сокращению.
Первыми при генетическом синдроме поражаются структуры, содержащие наибольшее количество важного белка – стенки кровеносных сосудов, связочный аппарат, цинновая связка глаза. Изменённая соединительная ткань не способна выполнять своей функции, выдерживать физическую нагрузку в связи с потерей прочности и упругости, у ребёнка возникают симптомы заболевания
Недуг относится к генетическим и передаётся от родителей по аутосомно-доминантному типу. Риск появления малыша с наследственным синдромом очень высокий, если у мамы или папы имеются признаки болезни. В 75% случаях заболеваний прослеживается появление недуга в каждом поколении семьи. У 25% больных определяется новая, спонтанная мутация, не находится чёткой связи с наследованием.
Соединительная ткань не образует отдельного органа в человеческом теле. Но её клетки располагаются во всём организме. По средствам этих структур выполняются опорная, защитная и трофическая функции, образуется своеобразный каркас и покровы всех органов. К разновидностям соединительной ткани относят хрящевую, костную, мышечную, жировую ткани, кровь и лимфу. Поэтому системные заболевания, связанные с тканевой патологией, отличаются большим многообразием проявлений.
Диагностика
Диагностика заболевания основана на анализе родословных, генетических тестах, поиска лица, с которого началось заболевание (пробанда), наличия симптомов синдрома Марфана.
При постановке диагноза у ребенка педиатр изучает его физическое и нервно-психическое развитие, состояние отдельных органов и систем.
Оценка состояния организма проводится с помощью ряда фенотипических диагностических тестов, теста на арахнодактилию, определения индекса Дюранта-Лайнера, индекса телосложения Варги.
Для постановки диагноза необходимо наличие одного из пяти основных симптомов и двух из дополнительных.
Методы терапии
Прежде всего, лечение заключается:
- в назначении бета-адреноблокаторов;
- в хирургическом вмешательстве при патологиях клапанов и аорты;
- в хирургической коррекции патологий позвоночника.
Из бета-блокаторов пациентам целесообразно назначать препараты в виде пропранолола или атенолола. Это помогает предотвратить серьёзные осложнения со стороны сосудов и сердца. Бета-блокаторы уменьшают интенсивность сокращения сердечной мышцы, снижая её нагрузку, останавливают процесс расслоения аорты и снижают риск развития аневризмы. Если аневризма достигает критических размеров, пациентам показано хирургическое вмешательство.
В качестве консервативного лечения сколиоза, обычно, применяют фиксацию позвоночника, но если он искривлён от 40 градусов и более, операция является более предпочтительным методом.
Всем пациентам, страдающим синдромом Марфана, нужно каждый год проходить обследование у невролога, кардиолога и окулиста, с генетическим консультированием по показаниям.
Что происходит при синдроме Марфана?
У всех есть белок фибриллин-1, который образует эластичные волокна в соединительной ткани. Фибриллин-1 также влияет на другой белок в вашем организме, трансформируя бета-фактор роста (TGF-бета), который помогает контролировать рост и развитие организма. Люди с синдромом Марфана наследуют мутацию гена, которая изменяет то, как организм использует фибриллин-1, что приводит к избытку факторов роста, это вызывает:
- Ткани кровеносных сосудов, сердца, связок, сухожилий и хрящей растягиваются сильнее, что делает их слабыми.
- Необычно сильный рост костей, делая их длиннее, чем обычно.
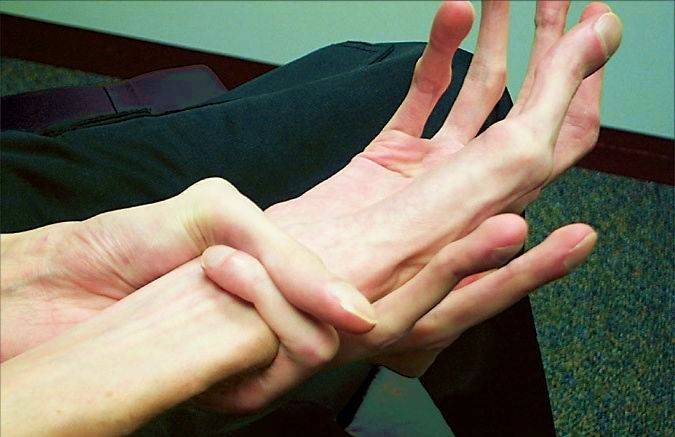
Проявления заболевания
Данной патологии сопутствует множество нарушений в организме. Так называемая «триада Марфана» — это повреждение скелетных костей, болезни сердечно-сосудистой системы и различные нарушения зрения. Однако надо отметить, что на умственные способности этот синдром никак не влияет.
Заболевшие отличаются, как правило, непропорциональным сложением: при довольно высоком росте у них укороченное туловище, длинные руки и пальцы, напоминающие паучьи (арахнодактилия), астеническое телосложение с мышечной гипотонией, деформация грудной клетки, сильное искривление позвоночника (кифоз, сколиоз, подвывихи и вывихи шейного отдела), плоскостопие.
Синдром Марфана, сопровождающийся аномалией сердечно-сосудистой системы, выражается в дефектах стенок крупных сосудов, особенно аорты и больших ветвей легочных сосудов, пороками развития сердца. Эти изменения часто формируются уже у развивающегося плода. Самая опасная форма синдрома Марфана, симптомы которой проявляются уже при рождении, приводит к прогрессирующей сердечной недостаточности и летальному исходу на первом году жизни младенца.
3.Симптомы и диагностика
Синдром Марфана обнаруживается уже в раннем возрасте; он может прогрессировать или оставаться относительно стабильным. Классической триадой считают деформацию опорно-двигательного аппарата, офтальмологическую патологию и сердечнососудистую недостаточность.
Больные отличаются высоким ростом и диспластичным телосложением: непропорционально длинные конечности, «паучьи пальцы» (арахнодактилия), килевидная или вдавленная грудная клетка, сложные искривления позвоночника (лордоз, кифосколиоз), вальгусная или варусная конфигурация ног, патологическая подвижность суставов, плоскостопие и т.д.
Характерные диспропорции обнаруживаются также в строении челюстно-лицевых структур. Явно недостаточно развиты мышечная и жировая ткани. Типичным признаком является аневризма аорты и/или других крупных сосудов, в т.ч. легочных и мозговых; встречается множество различных пороков сердца. Столь же полиморфными и тяжелыми могут быть аномалии развития органов дыхания, пищеварения, а также центральной нервной и зрительной систем (в частности, офтальмологически выявляется выраженная миопия или гиперметропия, подвывих хрусталика, полное отсутствие радужки или хрусталика, удлинение глазного яблока и т.п.).
Комплекс столь критичных по сути аномалий может быть выражен относительно мягко, однако в целом синдром Марфана обычно нуждается в квалифицированном терапевтическом наблюдении и сопровождении, – в противном случае и качество, и продолжительность жизни резко ограничиваются (в отсутствие курации непосредственной причиной смерти становится, как правило, хроническая или острая, наступающая вследствие разрыва аневризмы, сердечная недостаточность).
Диагноз устанавливается путем изучения семейного анамнеза и всестороннего обследования: практически любой из существующих сегодня диагностических методов выявляет те или иные характерные изменения.
Теги
Консультация детского Консультации в Консультация детского Консультации в Консультация генетикаПервичные консультации детских Консультация детского Консультации в Консультация кардиолога Консультация офтальмолога Врачи Врачи постоянным врачебным наблюдением Врачи и врачам Врачи к врачам пок врачам.у врачей различныху врачей пациентам просмотров Лечение при синдроме Причины синдрома Прогноз сопровождающееся преимущественным поражением Причины синдромафибриллина при синдромеМарфана приводят кпотере прочности итакже присутствием мутацийпрофилактические осмотры дляна осмотре упроведет осмотр сплановом осмотре врачмедицинском осмотре и осмотр глазмедицинского осмотра без осмотра вПервичный осмотр ипроходить осмотры у
врачиосмотраортопедомузимедицинскихсанктпетербургктнаукцентртравматологазаписатьсятелефонтикатегорииколичествоотделениевысшейрентгенпрофессорклиникиместесреднейклассацентральнаякандидатдокторомкрасногвардейский
Почему при определении признаков синдрома Марфана нужно обратиться к врачу?
Сама по себе генетическая аномалия совместима с жизнью. Однако опасны последствия болезни, вызванной FBN1 мутацией:
- разрывы крупных сосудов, чаще всего — аорты;
- хроническая сердечная недостаточность — неспособность сердца обеспечивать необходимую работу для кровоснабжения всех органов;
- снижение остроты зрения или полная потеря зрительной функции.
Разрыв аневризмы аорты или другого магистрального сосуда часто заканчивается моментальным летальным исходом. Хроническая сердечная недостаточность может перейти в острую форму, а без экстренной медицинской помощи также привести к фатальным последствиям — внезапной коронарной смерти. Именно эти осложнения чаще всего приводит к гибели детей с синдромом Марфана. Особая опасность ждет женщину с синдромом мутации гена FBN1 во время беременности: повышенная нагрузка на аорту в разы увеличивает риск ее разрыва.
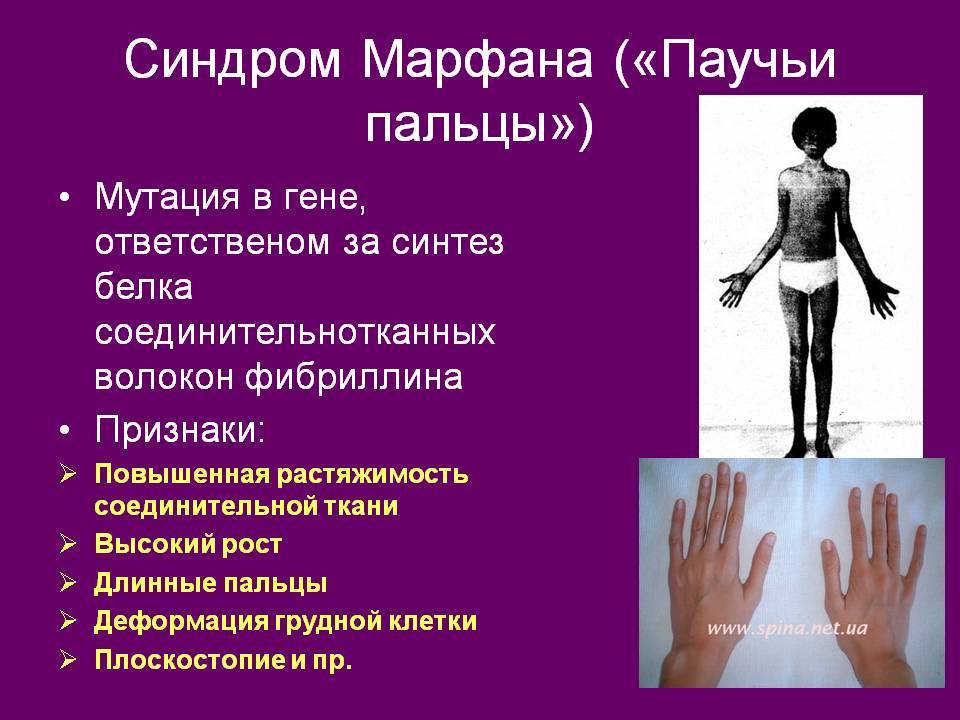


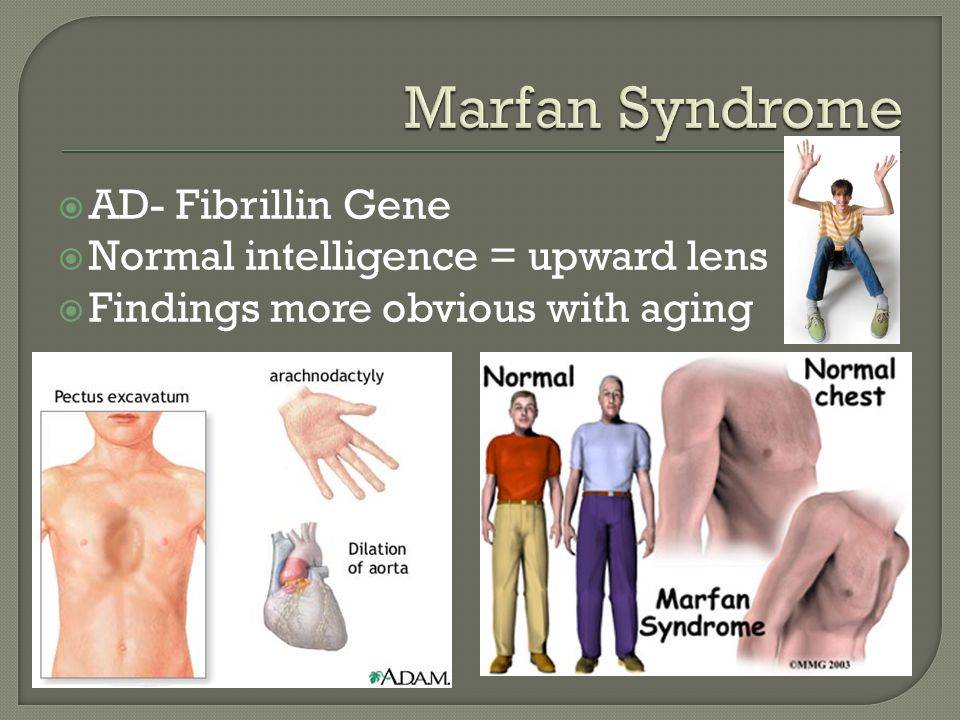


Чтобы предупредить развитие опасных осложнений и компенсировать возникающие нарушения, родителям нужно как можно раньше обратиться за медицинской помощью при первом подозрении на синдром Марфана у ребенка
При этом важно не только однократно провести обследование, но и стать на учет к врачам, которые занимаются коррекцией проявлений синдрома:
- специалисту по генетическим болезням;
- кардиологу;
- ортопеду-вертебрологу;
- дерматологу;
- офтальмологу;
- гастроэнтерологу.
Список специалистов зависит от степени выраженности заболевания, при этом регулярно необходимо проходить комплексные профилактические осмотры для раннего выявления новых нарушений.
Синдром Марфана — болезнь гениев?
С синдромом Марфана связаны не только многочисленные поводы для обращения к врачам. Часто люди с мутацией гена FBN1 компенсируют физические проявления болезни интеллектуальными способностями, поэтому это генетическое заболевание даже называют «синдромом гениев». Считается, что повышенный выброс адреналина из-за патологических изменений в надпочечниках определяет высокий тонус умственной и психической активности у таких пациентов. Именно поэтому в числе людей с синдромом Марфана можно найти известных личностей. Например, Юлию Цезарю, Аврааму Линкольну и Шарлю де Голлю патология не помешала стать известными политическими деятелями; Ганс Христиан Андерсен и Корней Чуковский создали уникальные литературные произведения, а Никколо Паганини прославился как гениальный музыкант.
Современные знаменитости также не скрывают свои недостатки и становятся еще более популярными из-за генетического дефекта. Например, солисту американской рок-группы Deerhunter Брэдфорду Коксу нетипичная внешность придает особый шарм, а испанский актер Хавьер Ботет очень востребован, поскольку правдоподобно и талантливо играет отрицательных героев в голливудских фильмах ужасов (рис. 6).